本文来源于中国科技大学计算机科学与技术学院刘淇教授课题组于2023年发表在ICLR2023上的文章《MOLECULE GENERATION FOR TARGET PROTEIN BINDING WITH STRUCTURAL MOTIFS》。

在本文中,作者提出了一个基于片段的分子生成网络,FLAG (Fragment based LigAnd Generation framework)。在FLAG中,从数据集中提取共同的分子片段,构建了motif的词汇库。在每个生成步骤中,首先采用 3D 图神经网络对中间上下文信息(口袋)进行编码。 然后,FLAG模型选择中心motif,预测下一个motif类型,并连接motif。其中,键长/键角通过化学信息学工具快速准确地确定。 最后,根据预测的旋转角度和结构细化进一步调整分子几何形状。
本文是为数不多基于片段的分子生成技术,且能提供分子的3D结合pose。
一、文章主要结果
分子生成过程示例如下图:

motif的提取过程如下图:

作者评估的结果显示,FLAG模型在生成分子的QED,SA,docking score以及分子的真实性,特别是C-O键等二面角分布上,具有明显的优势。
分子性质结果如下图:

生成分子的docking 情况:生成的分子结构正常,且能合理占据口袋,docking score的打分与原始参考分子相近。

生成分子真实性(二面角):

作者提供了开源代码:https://github.com/zaixizhang/FLAG
二、环境安装与数据准备
首先,从GitHub中复制项目:
git clone https://github.com/zaixizhang/FLAG.git项目目录如下:

然后根据yaml环境文件,安装conda 环境然后进行激活,因为torch和NVCC都是常见的版本,所以安装非常简单:
然后根据yaml环境文件,安装conda 环境然后进行激活,因为torch和NVCC都是常见的版本,所以安装非常简单:
conda env create -f flag_env.yaml
conda activate flag_env然后进入./data目录,创建FLAG文件夹(该文件夹必须要创建,作者提供的代码使用了该目录,但是开源代码中并未创建),按照README.md说明从谷歌云盘上下载数据,并保存到 ./data/FLAG 中(链接:https://drive.google.com/drive/folders/1CzwxmTpjbrt83z_wBzcQncq84OVDPurM)

然后,进行解压:
tar -xzvf crossdocked_pocket10.tar.gz数据预处理,motif词汇库的构建:
cd utils
python mol_tree.py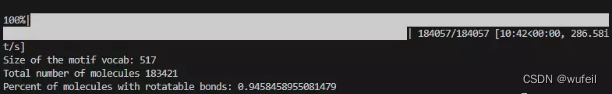
从上图可知,一共提取了517个motif,分子总数为183421个。运行结束以后,会在./data目录中,生成crossdocked_pocket10_processed.lmdb和crossdocked_pocket10_processed.lmdb-lock数据文件,./data目录如下:
同时,./utils目录下会生成语motif的语料库:vocab.txt。
注意,mol_tree.py文件有文件路径修改:
index_path = '/data/FLAG/crossdocked_pocket10/index.pkl' # wufeilwith open(index_path, 'rb') as f:index = pickle.load(f)for i, (pocket_fn, ligand_fn, _, rmsd_str) in enumerate(tqdm(index)):if pocket_fn is None: continuetry:path = '/data/FLAG/crossdocked_pocket10/' + ligand_fn # wufeilmol = Chem.MolFromMolFile(path, sanitize=False)moltree = MolTree(mol)cnt += 1if moltree.num_rotatable_bond > 0:rot += 1except:continue改为:
index_path = '../data/FLAG/crossdocked_pocket10/index.pkl' # wufeilwith open(index_path, 'rb') as f:index = pickle.load(f)for i, (pocket_fn, ligand_fn, _, rmsd_str) in enumerate(tqdm(index)):if pocket_fn is None: continuetry:path = '../data/FLAG/crossdocked_pocket10/' + ligand_fn # wufeilmol = Chem.MolFromMolFile(path, sanitize=False)moltree = MolTree(mol)cnt += 1if moltree.num_rotatable_bond > 0:rot += 1except:continue三、训练FLAG模型
python train.py如果直接运行,上述训练代码,会报:ModuleNotFoundErrons No module naned chemutils错误:
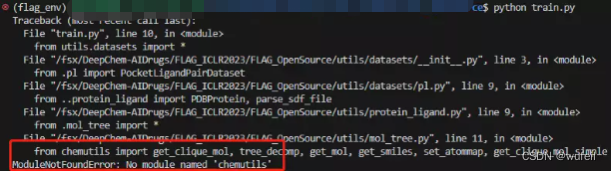
这是因为,代码路径问题,需要做如下修改,增加sys.path.append("./utils"):
import sys
sys.path.append("..")
sys.path.append("./utils") # wufeil同时,需要修改,训练的参数文件,即 ./configs/train_model.yml,修改内容如下,主要是调整了数据目录:

然后,python train.py可以正常运行,经过漫长时间的数据加载(> 1h),模型开始训练:
![]()
非常可惜,由于作者未提供训练好的checkpiont文件,所以需要我们自己先训练出一个模型,才能进行接下来的操作。
四、采样/分子生成
由于作者在GitHub上没有提供训练好的模型ckpt文件,因此只能使用我们自己训练的。在上一部训练结束以后,需要将模型的checkpoint 转移至 ./pretrained/并重命名为model.pt。然后执行如下命令:
python motif_sample.py脚本会调用./configs/sample_test.yml下的参数配置(如下),FALG模型会加载./test_data/project_1数据集,然后从中抽取第一个体系(代码是这么写的,而结果也是这样子的),进行分子生成。输入的结果保存在:/outputs/sample-0_2023_09_06__07_32_47。其中,sample-0_2023_09_06__07_32_47为采样的时间,生成的分子为sdf格式。从pocket_info.txt中可以查到口袋的信息,为BSD_ASPTE_1_130_0/2z3h_A_rec_1wn6_bst_lig_tt_docked_3_pocket10.pdb。
dataset:name: plpath: ./test_data/project_1model:checkpoint: ./pretrained/model.pthidden_channels: 256random_alpha: Truesample:seed: 2022num_samples: 100 # 每个配体/体系采样数量beam_size: 300logp_thres: -.infnum_retry: 5max_steps: 12batch_size: 10num_workers: 4在./configs/sample_test.yml配置文件中,设置的每个体系是采样100个,但是生成的sdf文件只有39个,说明不是每一次尝试sampling都是成功的。然后,我们将生成的分子放回到口袋中,几个示例如下:



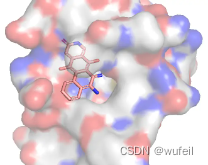
从上述结果来看,在可以被保存成sdf的分子中,还有很多分子是不合法的。但是比较严重的是,在生成分子中,出现了大量的非常复杂的并环,这些分子的复杂程度根本就不可能合成。
可能是因为,训练不足导致的,这里使用的参数文件是经过了18000次迭代的。所以,模型的真实结果还是需要作者提供原始的ckpt文件才能知道。但是,并环在FLAG模型中,容易生成,可能是真实的,作者给出的几个案例,全部都有并环的存在,如下图:

但,必须指明的是,这不意味着作者的工作存在严重缺陷,作为一种方法的验证,作者开辟了基于分子片段的分子生成技术,是非常有参考意义的。GitHub中的代码,更是具有借鉴意义。
五、为自定义的案例生成分子
在1.2中,作者提供的是批量的是生成分子,利用的是数据预处理完成后的pl数据集中test集,然后对其中的第一个数据进行分子采样,并没有提供一个分子从数据预处理开始。为了使用自己的例子进行分子生成,新编写了一个generate_mols.py脚本。
执行: python generate_mols.py 即可。
脚本会默认使用./utils/vocab.txt片段库,同时将生成的分子保存在./tests。
generate_mols.py 会调用./configs/sample_test.yml配置文件,文件内容如下:
dataset:name: plpath: ./test_data/project_1 model:checkpoint: ./pretrained/model.pthidden_channels: 256random_alpha: Truesample:seed: 2022num_samples: 50 # 每个配体/体系采样数量,至少要大于批次大小beam_size: 300logp_thres: -.infnum_retry: 5max_steps: 12batch_size: 10num_workers: 4其中,dataset中name必须为pl,因为原文作者设置如果数据集名字不为pl会报错。path为我们需要进行分子生成的体系保存路径,里面包含了两个体系,分别作为一个文件夹,目录如下:
./test_data/project_1
├── 3tym
│ ├── 3tym_ligand.sdf
│ └── 3tym_protein.pdb
├── 5tbo
│ ├── 5tbo_ligand.sdf
│ └── 5tbo_protein.pdb
└── index.pklmodel和sample字段下为模型相关的内容,没有做改变。sample中num_samples可以做调整,但是要大于batch_size。
在示例中,我们进行测试的两个体系是3tym和5tbo,生成的结果目录./test如下:
./tests
└── sample_test-pl_2023_09_13__02_10_37├── 3tym│ ├── 17.sdf│ ├── 27.sdf│ ├── 37.sdf│ ├── 47.sdf│ ├── SMILES.txt│ └── pocket_info.txt├── 5tbo│ ├── 19.sdf│ ├── 39.sdf│ ├── 47.sdf│ ├── 49.sdf│ ├── 9.sdf│ ├── SMILES.txt│ └── pocket_info.txt├── log.txt└── sample_test.yml虽然设置每个体系采样50个,但是真正生成的有小分子并不多,分别只有
5tbo生成分子的结果示例:


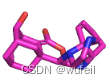
3tym生成分子的结果示例:
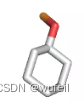


实在是因为我们训练迭代的次数太低,结果真的没法看。如果要用FLAG真的为靶点进行分子生成,显然需要进行更多次的迭代。可能数据集以及分子的切割方式也要做改进。