CAFE(Computational Analysis of gene Family Evolution)是一款以解释系统发育历史的方式分析基因家族大小变化的软件,这种分析常被称为基因家族收缩扩张(Gene family expansions and contractions)分析。
CAFE使用出生和死亡过程来模拟用户指定的系统发育树中的基因获得和丢失,可计算由父节点到子节点的基因家族大小转移率,也可推断祖先物种的基因家族大小,在该模型下生成的基因家族规模分布可以为评估观察到的类群之间家族规模差异的显著性提供基础。
自2005年Hahn课题组提出评估基因家族进化速度和模式的算法,2006年第一个版本CAFE发表后,2020年推出了最新版本CAFE5,之前旧版本的基本模型假设所有基因家族都具有相同的进化速率。新版本支持伽马分布速率类别对家族之间的速率变化进行显式建模。2020年发表,相较于cafe4来讲操作更加方便并且新增了模型(Gamma)。
CAFE5下载
1.安装
git clone https://github.com/hahnlab/CAFE5.git
cd CAFE5
./configure
make2.cafe5的使用
输入文件至少要两个,一个是基因家族数目统计文件Genefamilies_Count.tsv,一个是树文件tree.txt(带有分化时间),还可以增加一个lambda文件
2.1 常用参数
--infile, -iPath to tab delimited gene families file to be analyzed - Required for estimation.--n_gamma_cats, -kNumber of gamma categories to use. If specified, the Gamma model will be used to run calculations; otherwise the Base model will be used.--pvalue, -PP-value to use for determining significance of family size change, Default=0.05.--tree, -tPath to file containing newick formatted tree - Required for estimation.--lambda_tree, -yPath to lambda tree, for use with multiple lambdas.--output_prefix, -oOutput directory - Name of directory automatically created for output. Default=results.2.2 输入文件准备
2.2.1 数据准备
2.2.1.1识别基因家族:
(1)仅保留每个基因中有代表性的转录本,去除可变剪切和冗余基因;
(2)建立BLAST数据库,使用blastp进行 all-by-all 的比对;
(3)使用MCL基于blastp结果进行聚类,基因序列相似的通常是一个基因家族;
(4)解析MCL的输出结果,用作CAFE的输入。
(1)将所有最长的转录本合并成单个文件
#提取每个基因中最长的转录本
python python_scripts/cafetutorial_longest_iso.py -d twelve_spp_proteins/
#合并每个物种的最长转录本成为1个文件
cat twelve_spp_proteins/longest_*.fa | /data1/spider/ytbiosoft/seqkit rmdup - > makeblastdb_input.fa
(2)All-by-all BLAST
##我的blast工具安装在miniconda的一个子环境里面,首先需要激活这个环境
source/data1/spider/miniconda3/bin/activate
conda activate bioinforspace##先用makeblastdb建立BLAST数据库
makeblastdb -in makeblastdb_input.fa -dbtype prot -out blastdb##之后用blastp进行序列搜索,得到每个序列的相似序列
blastp -num_threads 20 -db blastdb -query makeblastdb_input.fa -outfmt 7 -seg yes > blast_output.txt &
#参数:
-seg参数过滤低复杂度的序列(即氨基酸编码为X)
-num_threads线程数,此处设置为20
(3)使用MCL进行序列聚类
根据BLAST输出中序列相似性信息寻找聚类,这些聚类将用于后续CAFE分析的基因家族。
##使用shell命令将BLAST转成MCL能够识别的ABC格式grep -v "#" blast_output.txt | cut -f 1,2,11 > blast_output.abc##创建网络文件(.mci)和字典文件(.tab),然后进行聚类
source /data1/spider/miniconda3/bin/activateconda activate orthofinder ##mcxload安装在orthofinder环境中
mcxload -abc blast_output.abc --stream-mirror --stream-neg-log10 -stream-tf 'ceil(200)' -o blast_output.mci -write-tab blast_output.tab &#参数:
--stream-mirror: 为输入创建镜像,即每一个X-Y都有一个Y-X
--stream-neg-log10: 对输入的数值做-log10 转换
-stream-tf: 对输入的数值进行一元函数转换,这里用到的是ceil(200)
#根据mci文件进行聚类, 其中主要调整的参数是-I,它决定了聚类的粒度,值越小那么聚类密度越大。一般设置为3。
mcl blast_output.mci -I 3
mcxdump -icl out.blast_output.mci.I30 -tabr blast_output.tab -o dump.blast_output.mci.I30
(4)整理MCL的输出结果
上一步MCL的输出还不能直接用于CAFE,还需要对其进行解析并过滤。
##1.将原来的mcl格式转成CAFE能用的格式
python python_scripts/cafetutorial_mcl2rawcafe.py -i dump.blast_output.mci.I30 -o unfiltered_cafe_input.txt -sp "ENSG00 ENSPTR ENSPPY ENSPAN ENSNLE ENSMMU ENSCJA ENSRNO ENSMUS ENSFCA ENSECA ENSBTA"
##2.将那些基因拷贝数变异特别大的基因家族剔除掉,因为它会造成参数预测出错。以下脚本是过滤掉一个或多个物种有超过100个基因拷贝的基因家族。
python python_scripts/cafetutorial_clade_and_size_filter.py -i unfiltered_cafe_input.txt -o filtered_cafe_input.txt -s
##3.将原本的编号替换成有意义的物种名
sed -i -e 's/ENSPAN/baboon/' -e 's/ENSFCA/cat/' -e 's/ENSBTA/cow/' -e 's/ENSNLE/gibbon/' -e 's/ENSECA/horse/' -e 's/ENSG00/human/' -e 's/ENSMMU/macaque/' -e 's/ENSCJA/marmoset/' -e 's/ENSMUS/mouse/' -e 's/ENSPPY/orang/' -e 's/ENSRNO/rat/' -e 's/ENSPTR/chimp/' filtered_cafe_input.txt
sed -i -e 's/ENSPAN/baboon/' -e 's/ENSFCA/cat/' -e 's/ENSBTA/cow/' -e 's/ENSNLE/gibbon/' -e 's/ENSECA/horse/' -e 's/ENSG00/human/' -e 's/ENSMMU/macaque/' -e 's/ENSCJA/marmoset/' -e 's/ENSMUS/mouse/' -e 's/ENSPPY/orang/' -e 's/ENSRNO/rat/' -e 's/ENSPTR/chimp/' large_filtered_cafe_input.txt
2.2.1.2物种树推断
构建物种树主要分为多序列联配和系统发育树两步,随后,在已有进化树的基础上构建超度量树用作CAFE的输入。
①多序列联配一般用的是单拷贝的直系同源基因,分别进行多序列联配,然后合并成单个文件;
②使用系统发育树推测软件进行建树(软件: RAxML, PhyML, FastTree; MrBayes),生成的系统发育树用NEWICK格式保存为twelve_spp_raxml_cat_tree_midpoint_rooted.txt;
cat twelve_spp_raxml_cat_tree_midpoint_rooted.txt
(((cow:0.09289 ,(cat:0.07151,horse:0.05727):0.00398):0.02355,((((orang:0.01034,(chimp:0.00440,human:0.00396):0.00587):0.00184,gibbon:0.01331):0.00573,(macaque:0.00443,baboon:0.00422):0.01431):0.01097,marmoset:0.03886):0.04239):0.03383,(rat:0.04110,mouse:0.03854):0.10918);
③推断超度量树
超度量树(ultrametric tree)也叫时间树,就是将系统发育树的标度改成时间,从根到所有物种的距离都相同。构建方法有很多,比较常用的就是r8s。详情参考文章
#使用cafetutorial_prep_r8s.py构建r8s的批量运行脚本,然后提取超度量树
python python_scripts/cafetutorial_prep_r8s.py -i twelve_spp_raxml_cat_tree_midpoint_rooted.txt -o r8s_ctl_file.txt -s 35157236 -p 'human,cat' -c '94'
/data1/spider/liupiao/biosoft/r8s1.81/src/r8s -b -f r8s_ctl_file.txt > r8s_tmp.txt &
tail -n 1 r8s_tmp.txt | cut -c 16- > twelve_spp_r8s_ultrametric.txt2.2.2. Genefamilies_Count.tsv
制表符分隔的基因家族计数文件,通常用OrthoMCL,
OrthoFinder等软件获取计数信息。
示例格式
Desc Family ID human chimp orang baboon gibbon macaque marmoset rat mouse cat horse cow
ATPase ORTHOMCL1 52 55 54 57 54 56 56 53 52 57 55 54
(null) ORTHOMCL2 76 51 41 39 45 36 37 67 79 37 41 49
HMG box ORTHOMCL3 50 49 48 48 46 49 48 55 52 51 47 55
(null) ORTHOMCL4 43 43 47 53 44 47 46 59 58 51 50 55
Dynamin ORTHOMCL5 43 40 43 44 31 46 33 79 70 43 49 50
......
....
..
DnaJ ORTHOMCL10016 45 46 50 46 46 47 46 48 49 45 44 48
我们首先利用OrthoFinder的Orthogroups.GeneCount.tsv文件生成符合要求的输入文件
cp Results_May02/Orthogroups/Orthogroups.GeneCount.tsv CAFE/
awk 'OFS="\t" {$NF=""; print}' Orthogroups.GeneCount.tsv > tmp && awk '{print "(null)""\t"$0}' tmp > cafe.input.tsv && sed -i '1s/(null)/Desc/g' cafe.input.tsv && rm tmp
查看文件格式
Desc Orthogroup Aof.pro Ath.pro Atr.pro Cba.pro Cri.pro Csa.pro Csu.pro Kle.pro Mpo.pro Nco.pro Osa.pro Ppa.pro Smo.pro Tpl.pro Vca.pro Vvi.pro Zma.pro
(null) OG0000000 145 112 95 5 372 129 3 1 2 217 126 16 206 419 4 177 117
(null) OG0000001 9 4 3 1691 9 96 2 56 2 4 21 0 2 5 3 2 0
(null) OG0000002 32 117 62 1 92 117 2 0 20 81 119 77 40 193 5 107 161
(null) OG0000003 37 104 54 3 89 76 4 5 10 74 144 22 47 134 8 79 154
(null) OG0000004 73 104 51 4 40 80 2 10 12 76 87 33 22 136 5 97 135
(null) OG0000005 28 46 36 11 37 47 0 3 50 81 81 32 48 120 0 54 73
(null) OG0000006 41 43 74 6 38 57 0 4 25 57 52 19 33 155 0 87 40
(null) OG0000007 58 52 60 0 18 42 0 0 12 50 56 17 57 99 1 82 52
(null) OG0000008 38 57 26 7 52 47 4 6 19 40 59 43 20 29 1 41 80
(null) OG0000009 46 57 26 1 25 46 1 2 11 52 65 29 13 50 1 48 87
生成之后还需要剔除不同物种间拷贝数差异过大的基因家族,否则会报错,有内置脚本可以使用,我在运行的时候需要去掉第一行才能使用
python ~/soft/CAFE5/tutorial/clade_and_size_filter.py -i cafe.input.tsv -o gene_family_filter.txt -s 另一种方法:
awk 'NR==1 || $3<100 && $4<100 && $5<100 && $6<100 && $7<100 && $8<100 && $9<100 && $10<100 && $11<100 && $12<100 && $13<100 && $14<100 && $15<100 && $16<100 && $17<100 && $18<100 && $19<100 {print $0}' cafe.input.tsv >gene_family_filter.txt
最后的文件格式,保证第一行的物种名字与进化树的一致即可
Desc Orthogroup Aof Ath Atr Cba Cri Csa Csu Kle Mpo Nco Osa Ppa Smo Tpl Vca Vvi Zma
(null) OG0000020 26 37 23 4 35 28 0 1 24 28 43 24 27 47 0 35 42
(null) OG0000021 49 41 31 7 30 31 8 2 7 26 49 15 11 31 0 36 45
(null) OG0000022 27 25 31 0 27 34 2 1 23 25 46 18 27 44 1 39 45
(null) OG0000024 37 40 27 0 22 30 1 11 9 33 38 18 25 43 0 37 39
(null) OG0000029 28 26 23 2 24 25 1 2 5 32 34 31 17 35 1 30 40
(null) OG0000030 23 30 16 1 23 27 1 1 27 26 26 16 15 49 1 28 35
(null) OG0000031 28 36 26 3 27 23 8 1 3 17 37 10 18 38 3 34 28
(null) OG0000032 18 16 25 0 24 19 0 5 4 25 36 6 38 49 1 38 35
(null) OG0000033 19 17 20 0 12 16 4 6 18 35 42 4 23 39 3 45 28
(null) OG0000035 17 37 17 8 28 24 2 2 5 30 41 8 6 26 2 32 37
(null) OG0000036 22 15 17 3 22 19 7 12 13 20 24 20 30 38 3 35 22
(null) OG0000039 14 27 36 0 34 24 0 2 2 12 41 4 47 13 0 37 26
(null) OG0000040 15 30 9 1 19 35 0 2 11 25 26 19 12 48 0 39 272.2.2. tree.txt
本步骤直接使用mcmctree生成的FigTree.tre文件修改一下即可使用
grep "UTREE 1 =" FigTree.tre | sed -E -e "s/\[[^]]*\]//g" -e "s/[ \t]//g" -e "/^$/d" -e "s/UTREE1=//" > tree.txt
3.1 运行CAFE5
cafe5 -i gene_family_filter.txt -t tree.txt -o out -c 1
## 如果使用Gamma模型与泊松分布cafe5 -i gene_family_filter.txt -t tree.txt -o out -c 1 -k 2 -p ##注意-k可以调,一般为2-5
3.2 CAFE5输出结果
3.2.1 结果文件
Gamma_asr.tre ## 每个基因家族的树文件
Gamma_branch_probabilities.tab ## 每个分支计算的概率
Gamma_category_likelihoods.txt
Gamma_change.tab ## 每一个基因家族在每个节点的收缩与扩增数目
Gamma_clade_results.txt ##每个节点基因家族的扩增/收缩数目
Gamma_count.txt ## 每一个基因家族在每个节点的数目
Gamma_family_likelihoods.txt
Gamma_family_results.txt ## 基因家族变化的p值和是否显著的结果
Gamma_results.txt ## 模型,最终似然值,最终Lambda值等参数信息。
注释:我们主要用的文件有Gamma_asr.tre(主要对应后面表格中的节点)、Gamma_change.tab(看哪些基因家族在哪个节点发生变化)、Gamma_clade_results.txt(体现在树上,每个节点基因家族的收缩/扩增数目)、Gamma_family_results.txt(显著扩增/收缩的基因家族)
3.2.2 每个节点基因家族收缩/扩增数目的体现
其实有绘图脚本,但是很久没有更新,可能不适用于CAFE5,我们可以自己画
将基因家族的扩增/收缩数目体现在树上,需要两个文件,Gamma_asr.tre,Gamma_clade_results.txt
cat Gamma_clade_results.txt
#Taxon_ID Increase Decrease
Mpo<21> 232 1298
Ppa<20> 2231 371
<31> 134 65
<25> 949 220
<23> 134 209
Atr<13> 516 922
Kle<29> 493 741
<12> 245 56
<28> 314 340
<4> 118 176
Cri<17> 1669 287
<22> 214 184
<19> 445 93
<16> 291 352
Osa<3> 579 572
Aof<6> 935 840
<8> 142 112
Csa<1> 326 834
<7> 640 138
Tpl<15> 1147 395
<14> 204 273
Nco<11> 631 559
Zma<2> 1776 232
Vvi<5> 958 433
<10> 413 112
<9> 345 66
Smo<18> 842 1315
Cba<24> 744 1664
<30> 23 17
Ath<0> 1090 291
Csu<27> 305 1560
Vca<26> 438 1040
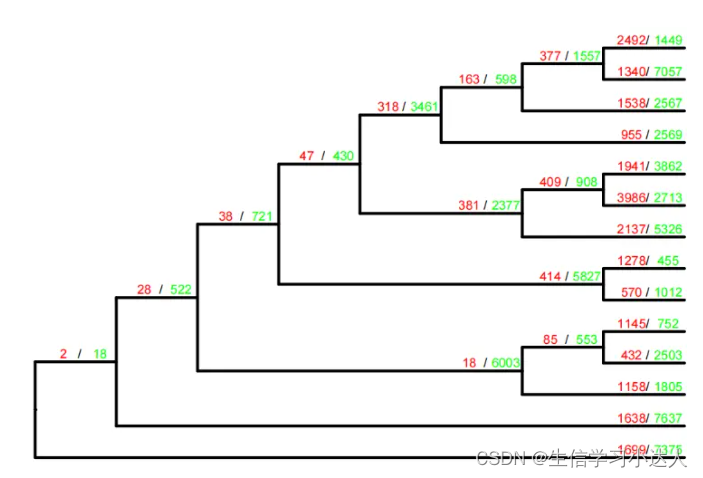
less Gamma_asr.tre
BEGIN TREES;TREE OG0000021 = ((Kle<29>*_2:820.007,(((Mpo<21>*_7:428.285,Ppa<20>*_15:428.285)<23>_12:70.3982,((Cri<17>_30:404.796,(Tpl<15>_31:308.175,(Atr<13>_31:208.47,(Nco<11>*_26:176.909,(((Osa<3>*_49:45.2652,Zma<2>_45:45.2652)<7>_45:60.4652,Aof<6>*_49:105.73)<9>*_44:25.8965,(Vvi<5>_36:101.378,(Csa<1>*_31:83.0358,Ath<0>*_41:83.0358)<4>_36:18.3421)<8>_36:30.2489)<10>*_36:45.2824)<12>_31:31.5605)<14>_31:99.7053)<16>*_30:96.6205)<19>*_27:48.1939,Smo<18>_11:452.99)<22>*_13:45.694)<25>*_12:190.787,Cba<24>_7:689.47)<28>*_7:130.537)<31>_5:177.864,(Csu<27>*_8:874.03,Vca<26>*_0:874.03)<30>_5:123.841)<32>_5
可视化:
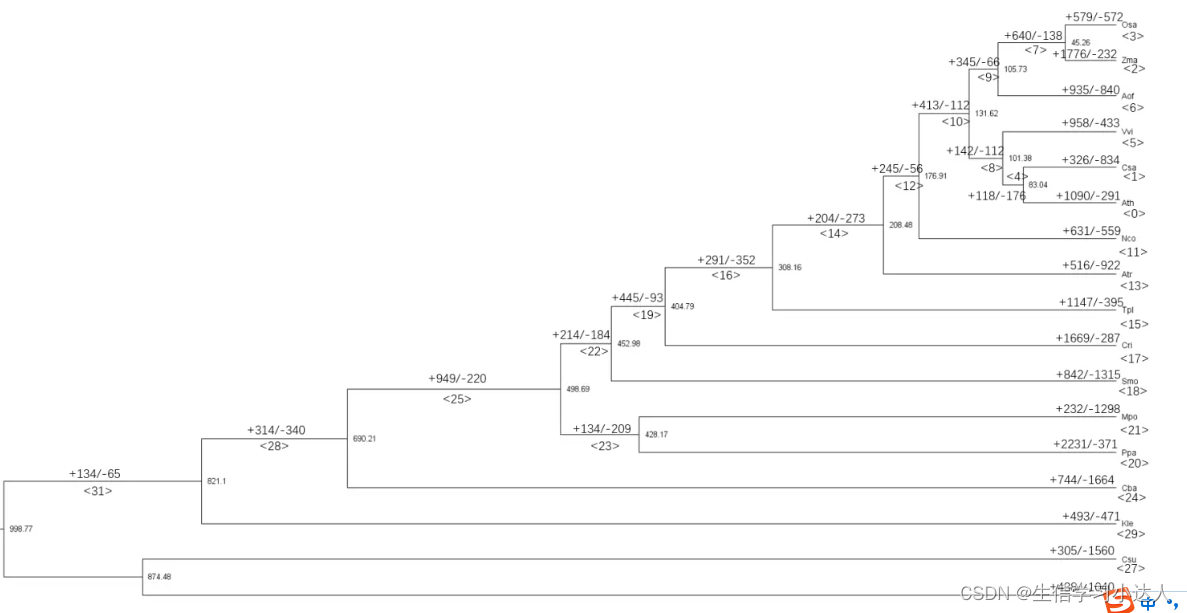
基因家族收缩和扩张
3.2.3 其他整理
相较于CAFE4,这些结果并没有直接体现显著扩张/收缩的基因家族,或者我们想找一下某个节点具体扩张的基因,可以结合目前拿到的输出文件进行进一步整理
cat Gamma_family_results.txt |grep "y"|cut -f1 >p0.05.significant
#提取显著扩张或收缩的orthogroupsID
grep -f p0.05.significant Gamma_change.tab > Gamma_p0.05change.tab
#显著扩张/收缩的基因家族在每个节点的收缩与扩增数目
cat Gamma_p0.05change.tab | cut -f1,2 | grep "+[1-9]" | cut -f1 > node0significant.expand
#Ath显著扩张的orthogroupsID
wc -l node0significant.expand
#Ath显著扩张的基因家族数目
cp ../../Results_May02/Orthogroups/Orthogroups.tsv ./
grep -f node0significant.expand Orthogroups.tsv | cut -f3 | sed "s/ /\n/g" | sed "s/\t/\n/g" | sed "s/,//g" | sort | uniq > node0significant.expand.genes
#提取Ath显著扩张的基因,方法一
cp ../../Results_May02/Orthogroups/Orthogroups.txt ./
grep -f node0significant.expand Orthogroups.txt |sed "s/ /\n/g" | grep "Ath" | sort | uniq > node0significant.expand.genes
#提取Ath显著扩张的基因,方法二
参考教程:
教程2
CAFE5下载安装与使用
利用cafe软件进行基因家族收缩与扩张分析
使用OrthoFinder进行直系同源基因分析
如何将cafes的结果可视化