| 欢迎浏览我的CSND博客! Blockbuater_drug …点击进入 |
|---|
文章目录
前言
分子对接是计算机辅助药物设计(CADD)中广泛使用的基本工具之一,主要过程可分为两部,首先是是探索到配体-受体结合的结合的正确姿势(pose identification),然后采用打分函数对其进行评估(pose Scoring)。
近年来,由于计算机硬件和数据集可用性的进步,数据驱动的机器学习方法已成为药物设计框架的重要组成部分之一。基于机器学习和深度学习构建打分函数,可以预测对接过程中生成的蛋白质-配体位姿或晶体复合物的结合得分,已成为CADD领域一个活跃的研究命题。
本文解读了一种先进的机器学习打分函数GB-Score的发展及原理,介绍相应验证环境的部署,以及如何使用,便于感兴趣的朋友作进一步优化。
一、GB-Score是什么?
GB-Score是一种最先进的基于机器学习的评分函数,利用包含23496组数据的PDBbind-v2019 general sets作为数据集,使用距离加权的原子间接触特征和梯度提升树算法来预测结合亲和力。
距离加权原子间接触特征化方法使用不同配体和蛋白质原子类型之间的距离来数值表示蛋白质-配体复合物。
GB-Score在CASF-2016基准测试中的得分能力指标非常优异,实现Pearson相关性0.862和RMSE 1.190。
Github代码:https://github.com/miladrayka/GB_Score
介绍文章:https://onlinelibrary.wiley.com/doi/10.1002/minf.202200135 原文自由下载:GB_Score.pdf
前期研究已经证明,配体和蛋白质原子之间的距离加权原子间接触可以作为机器学习过程中蛋白质-配体复合物数学表示的特征。本研究作者通过使用更好的特征选择、扩展的训练集和不同的学习算法来改进以前的评分函数ET Score。在特征选择上,采用了更合理的特征选择方法来缩小描述蛋白质-配体复合物的特征向量的尺寸,以变得更符合奥卡姆的剃刀规则。此外,在严格的环境中仔细检查新生成的评分函数GB Score,以评估其在新数据中的泛化能力。
对于配体,是基于元素划分原子类型(H、C、N、O、F、P、S、Cl、Br、I)。
对于蛋白质原子类型,根据侧链的化学性质将氨基酸残基分为四类(带电(c)、极性(p)、两亲性(a)、疏水性(h)),然后将相同的基于元素的原子类型归于每个组。通过这个分类,产生的蛋白质原子类型将反映蛋白质原子的局部化学环境。
下一步,计算特定原子类型对的所有原子间距离。大小低于预定义截止值(dcutoff)的距离由自然数(n)的逆幂加权并相加。之前的研究证明了12A和2分别是dcutoff和n的适当选择。对所有可能的原子类型对重复上述算法,并产生具有400维的特征向量作为蛋白质-配体复合物的表示。
在预处理步骤中,消除了所有静态、准静态(方差低于0.01)和相关(相关性高于95%)特征,这导致不同训练集的特征维度不同。此外,由于平均值和标准偏差,对其余特征进行了归一化。
数据集及处理后的特征向量维数Nf, 以及不同元素特征总和的权重,如下所示。
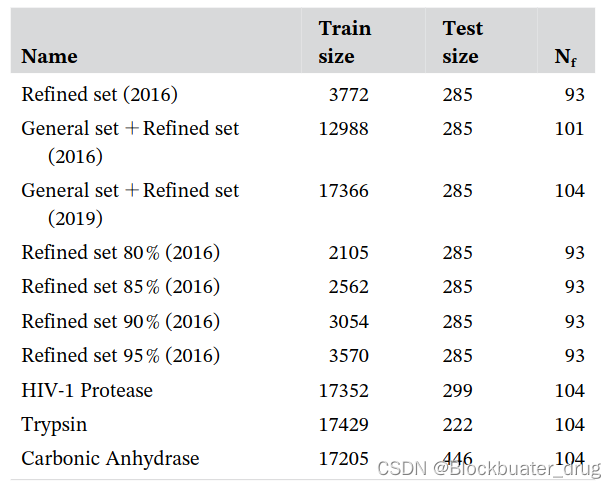
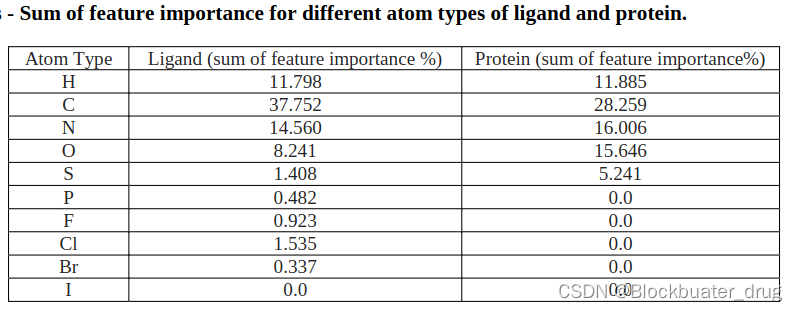
作者建立的三种机器学习算法RF,ERT,GBT,也是常见的机器学习算法类型。Scikit学习机器学习包用于训练。
在RF和ERT中,n_estiques设置为500,并且只有mtry(max_features)超参数做了优化。对于GBT,所有超参数都设置为参考论文中的值。
由于上述算法的随机性,训练过程重复十次,并且通过对十个以上的模型进行平均来报告模型的均方根误差(RMSE)和皮尔逊相关性(Rp)。相应的超参数如下所示:
以下是CASF2016(core sets)数据集上的打分效果,看起来已经相当不错。
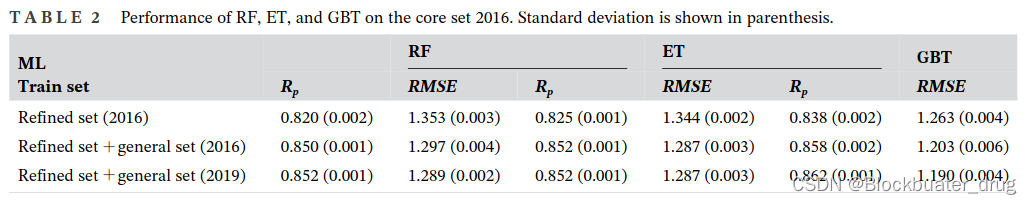 GBT算法整体优于其他两种算法的效果。
GBT算法整体优于其他两种算法的效果。
进一步在更大的数据集PDBbind 2019版的refined general sets上做了训练,Rp 和RMSE分别达到 0.862 和1.19,该模型被称为 GB-score。
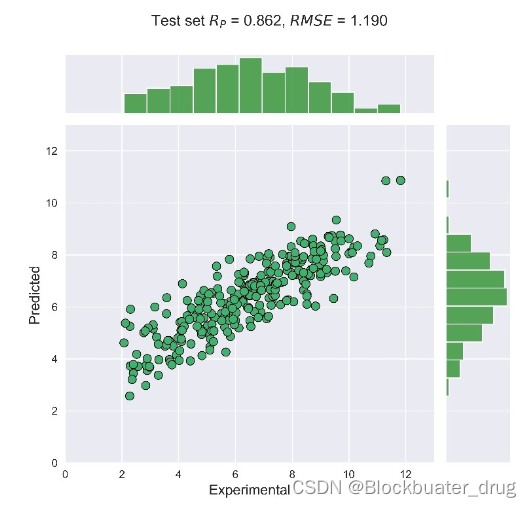
显示GB score预测了pKi/d值大于10的蛋白质配体结构的错误值。这一观察结果可归因于所使用的训练集,因为只有1.80%的训练集数据
具有大于10的pKa(Ka的负对数值),因此GB score预测偏向于中间范围pKi/d。所以,后续研究可以增加具有高pKi/d范围的数据作为解决方案。
为了验证模型的健壮性,在PDBbind 2019v数据集上做了5折交叉验证,Rp和RMSE分别为 0.764 (0.001) and 1.205 (0.007),有所下降,认为是数据数量增加和多样性增加所致。调整CASF2016数据集中的数量作为训练集,以及控制数据相似性均能证明GBT算法的稳健性。总之,即便测试集与训练集相似数据减少,机器学习算法仍然能很好的发挥作用。
core sets 2016分身按照蛋白家族分为57种,75%的家族预测结果相关系数可以达到0.7以上的可接受范围。
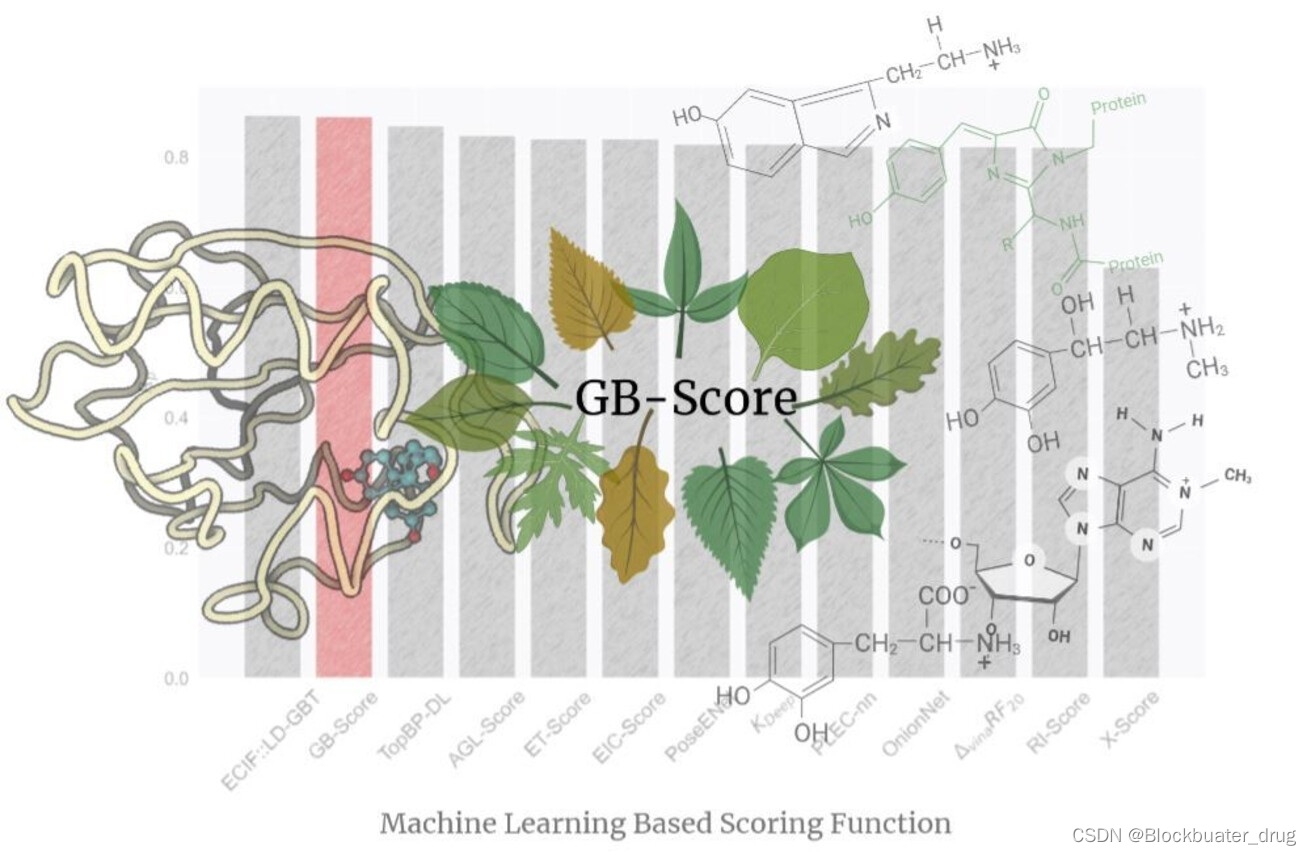
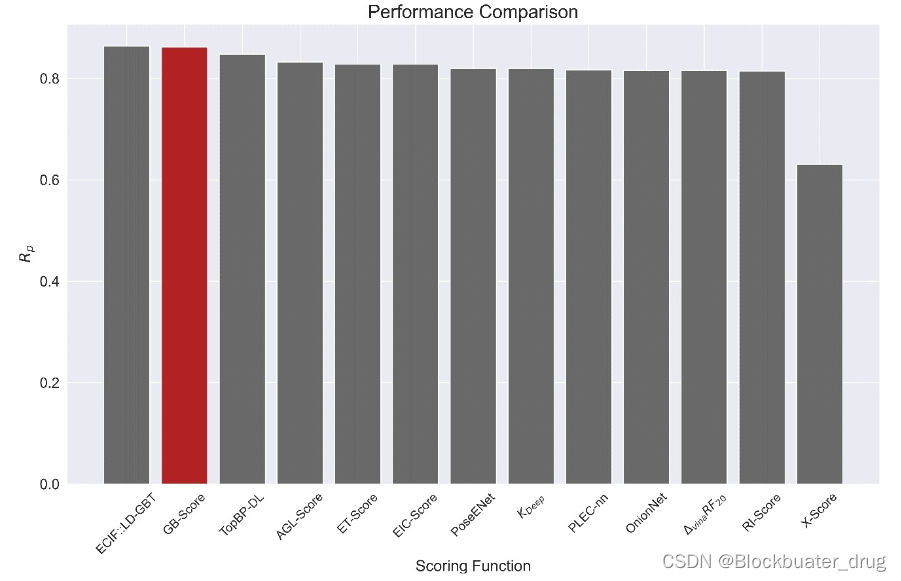
ECIF::LD-GBT,ECIF,AGL-Score ,ETScore ,EIC-Score,RosENet,KDEEP,PLEC-nn ,OnionNet ,DvinaRF20,RI-Score 和X-score. ECIF::LD-GBT表现最优Rp =0.866,GBscore表现次之(Rp =0.862).
不足与改进:更其他研究一样,缺少真实场景中的应用;而且打分函数本身依赖于对接pose。另外,应该完善机器学习打分函数的评估方法,目前的机器学习打分函数都得到非常接近的效果,难以辨别优劣,或许可以通过划分不同的适用范围来处理这个问题。
二、文献复现 -训练和验证环境
安装环境:Ubuntu 22.04。
1. GB score验证虚拟环境的配置
conda create -n gb_score_env python=3.8.8 numpy=1.21.2 pandas=1.2.4 seaborn=0.11.1 joblib=1.0.1 matplotlib=3.3.4
conda activate gb_score_env
python -m pip install biopandas==0.2.8 scipy==1.7.1 scikit-learn==0.24.1 progressbar2==3.53.1
conda install jupyter
2. Usage
1- Preparing ligand and protein file
a. Ligand and protein structure should be saved in .mol2 and .pdb format files respectively.
b. Each ligand and protein files for a specific complex must be placed in a same folder.
for example:
./1a1e/1a1e_ligand.mol2
./1a1e/1a1e_protein.pdb
./1a4k/1a4k_ligand.mol2
./1a4k/1a4k_protein.pdb
2- Generating features
运行generate_features.py 生成GB-Score的特征文件,类型为.csv文件:
-d 定义输入文件所在文件夹的路径; -f 定义输出特征的文件名。
python generate_features.py -d score/score_in/ -f feature.csv
3 - Repeat and extend current report
文件analysis.ipynb提供了复现整个设计及验证的过程,有明确的注释,感兴趣可以查看。
.csv 和.joblib文件需要提前下载解压到files和saved_model文件夹。
总结
本文解读了机器学习算法GB-Score的建立和验证工作。
机器学习算法有望显著提升对接pose打分的性能,但仍有多个方面问题亟待解决:
(1)缺少系统的机器学习打分函数评价方式;
(2)打分性能与对接pose质量紧密相关也是需要考虑的因素
(3)如何找到不同算法\不同原理打分函数的适用场景?
参考资料
- https://github.com/miladrayka/GB_Score
- https://onlinelibrary.wiley.com/doi/10.1002/minf.202200135
| 欢迎浏览我的CSND博客! Blockbuater_drug …点击进入 |
|---|