–https://doi.org/10.1093/bib/bbab573
A universal approach for integrating super large-scale single-cell transcriptomes by exploring gene rankings
打算深挖单细胞大模型的一系列文章、算法和代码,按时间线来去学习也许会好一些,所以第一篇带来的是2022年发表在Briefings in Bioinformatics的iSEEEK模型。
留意更多内容,欢迎关注微信公众号:组学之心
代码来源:https://github.com/lixiangchun/iSEEEK
数据基本情况和预处理
数据:scRNA-seq,11.9M / cross-tissue, cross-species(没公开说明具体用了什么数据);
数据处理:删除线粒体、核糖体和非编码蛋白质的基因。挑出126个top表达的基因作为每个单细胞的sentence(每个单细胞自己的top基因)。
一、模型架构
1个embedding layer + 8 个 encoder layers(每个encoder有576维度的 hidden units + 8 个 attention heads)带掩码操作,类似BERT模型。
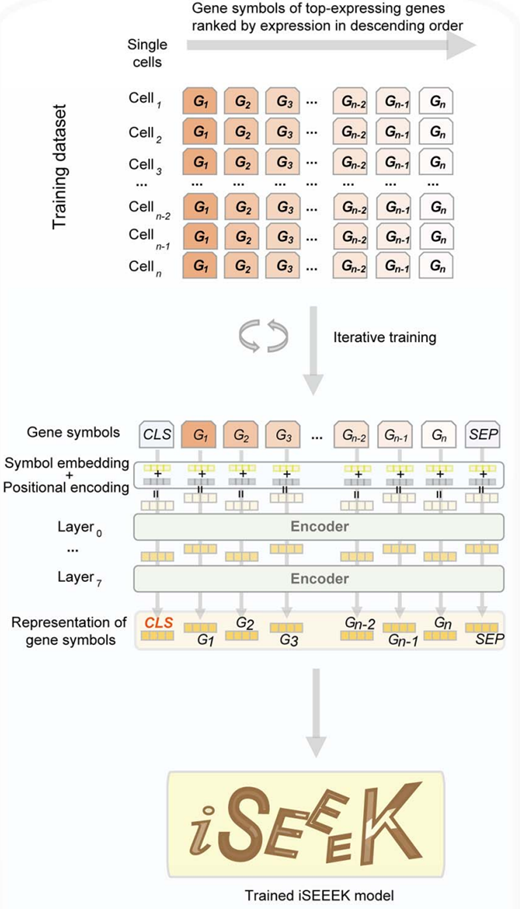
1.输入表示
文章用蛋白质编码的基因symbol构建了一本字典,一共20706个索引:

在过滤掉了细胞中表达极低的基因后,根据它们的表达水平进行排序。对每个细胞做一个128 个token的序列,其中token是基因symbol和特殊标记([CLS]、[SEP] 和 [PAD])。如果基因数量为 <126,将[PAD]标记填充到输入序列中。第一个token始终是[CLS],最后一个token始终是[SEP]。
2.Embedding layer
将 128 个基因symbol token和特殊标记的embedding,及位置encoding合起来(按照top的顺序来降序排列)作为输入。基因symbol首先被转换为索引。
3.Encoder layers
其中包含多头自注意力机制、前馈网络、层归一化层和残差连接。自注意力公式如下:
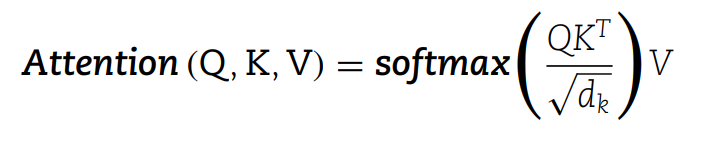
4.模型预训练
iSEEEK模型随机屏蔽输入中的一些基因,并根据它们的双向上下文预测masked基因的词汇索引。iSEEEK采用最大长度为126的基因symbol token作为输入。
训练过程中应用了与BERT相同的数据采样策略:training data generator随机选择15%的基因位置进行预测。如果选择了第 i 个基因,用
(1)80% 的time是 [MASK] 标记替换它,
(2)10% 的time是随机基因,
(3)10% 的time是原始不变的基因。
iSEEEK通过将其预测结果与原始基因进行比较,并用交叉熵损失进行训练。使用Adam的优化器,学习率为1e-4,β1 = 0.9,β2 = 0.999,前10000个步骤的学习率预热,学习率线性衰减,batch size=64,epoch=48。
二、模型训练主要代码
1.模型初始化
研究已经按照字典做好了tokenizer,上传到了Hugging face中。下载地址:https://huggingface.co/TJMUCH/transcriptome-iseeek/tree/main
tokenizer = PreTrainedTokenizerFast.from_pretrained("TJMUCH/transcriptome-iseeek")
model = BertForMaskedLM.from_pretrained("TJMUCH/transcriptome-iseeek")
运行transformer中PreTrainedTokenizerFast可以加载预训练分词器,from_pretrained 方法会自动下载和加载分词器配置、词汇表和其他必要资源。
运行BertForMaskedLM会读取TJMUCH/transcriptome-iseeek中的config.json文件,并使用其中的配置信息来初始化模型的架构和参数。这些配置包括模型的层数、注意力头的数量、隐藏层大小、最大位置编码、词汇表大小等。该文件的主要内容:
- _name_or_path:模型的来源或存储位置,显示是本地路径
- architectures: [“BertForMaskedLM”]:指定了模型的架构为 BertForMaskedLM
- attention_probs_dropout_prob:注意力机制中的 dropout 概率,0.1
- classifier_dropout:分类器的 dropout 概率,0.1
- gradient_checkpointing:是否启用梯度检查点,false
- hidden_act:隐藏层的激活函数类型,gelu
- hidden_dropout_prob:隐藏层的 dropout 概率,0.1
- hidden_size:隐藏层的维度,576
- initializer_range: 0.02,表示在 [-0.02, 0.02] 范围内初始化权重
- intermediate_size:Feed-forward 层的中间层大小,1536
- layer_norm_eps:LayerNorm 层中的 epsilon 值,用于防止除以零的数值稳定性,1e-12
- max_position_embeddings:最大位置编码数量,指模型可以处理的最大输入序列长度,384
- model_type:模型的类型,bert
- num_hidden_layers:模型包含 8 个 Transformer 层。
- num_attention_heads:每个层有 8 个注意力头。
- pad_token_id:填充标记的 ID,3
- position_embedding_type:位置编码的类型,“absolute”,表示使用绝对位置编码
- torch_dtype:指定模型权重的数值类型,float32
- transformers_version:使用的 transformers 库的版本
- type_vocab_size:词汇表的类型数量,2(用于句子对任务)
- use_cache:在解码过程中是否缓存计算结果,以加速推理
- vocab_size:模型的词汇表大小为 20,706,与 tokenizer.json 中定义的词汇表一致。
此外,还会下载预训练权重文件。最后初始化BertForMaskedLM模型。

2.加载数据:
文章的Github中有作者提供的训练集(1000个细胞),测试集(100个细胞),训练集长这样,1000行,每行有256个基因:

dataset, dataset_test, train_sampler, test_sampler, tokenizer = load_data(args.train_file, args.val_file, tokenizer, args.max_len, args.distributed)
来看看load_data自定义函数里面主要有啥:
dataset = GeneRankingDataset(train_file, tokenizer, max_len)
dataset_test = GeneRankingDataset(valid_file, tokenizer, max_len)class GeneRankingDataset(Dataset):def __init__(self, text_file, tokenizer, max_len):self.tokenizer = tokenizerself.max_len = max_lenself.lines = self.load_lines(text_file)def load_lines(self, text_file):lines = []f = open(text_file)for line in f:line = line.strip()if line.isspace():continuea = line.split()if len(a) <= self.max_len:lines.append(line)else:lines.append(" ".join(a[0:self.max_len]))f.close()return linesdef __len__(self):return len(self.lines)def __getitem__(self, idx):return self.tokenizer(self.lines[idx], add_special_tokens=True, truncation=True, padding=True, max_length=self.max_len)
GeneRankingDataset依照tokenizer的设定,处理输入数据每一行(load_lines)前128个基因(max_len=128),实际上是126个基因加上特殊标记头和尾。
接下来load_data函数代码是判断是否分布式训练(DistributedSampler),或者RandomSampler对训练集进行随机采样,SequentialSampler对测试集进行顺序采样:
print("Creating data loaders")
if distributed:train_sampler = torch.utils.data.distributed.DistributedSampler(dataset)test_sampler = torch.utils.data.distributed.DistributedSampler(dataset_test)
else:train_sampler = torch.utils.data.RandomSampler(dataset)test_sampler = torch.utils.data.SequentialSampler(dataset_test)
3.输入数据的预处理
加载完数据后,用transformer包的DataCollatorForLanguageModeling 方法处理语言模型的输入数据,还可以自动的为训练数据应用掩码。
collator在 DataLoader 中,每次从数据集中提取一批数据时,会将这些数据整理成适合模型输入的格式,尤其是批次化(batching)和掩码的应用。mlm_probability=0.15,设置掩码的概率,表示有多少比例的 token 会被随机选中并进行掩码处理。
15% 的 token 被选中进行掩码处理,但不是所有的被选中的 token 都会被替换为 [MASK]:
- 80% 的概率,将选中的 token 替换为 [MASK]。
- 10% 的概率,将选中的 token 替换为一个随机的 token。
- 10% 的概率,保持原 token 不变。
data_collator = DataCollatorForLanguageModeling(tokenizer=tokenizer, mlm=True, mlm_probability=0.15)data_loader = torch.utils.data.DataLoader(dataset, batch_size=args.batch_size, collate_fn=data_collator, sampler=train_sampler, num_workers=args.workers, pin_memory=True)data_loader_test = torch.utils.data.DataLoader(dataset_test, batch_size=args.batch_size, collate_fn=data_collator,sampler=test_sampler, num_workers=args.workers, pin_memory=True)
4.同步批量归一化 (SyncBatchNorm):
目的是在分布式训练中使用同步批量归一化。在分布式训练中,多个 GPU 可能会分别处理不同的数据子集。这时,如果各自的批量归一化层独立计算各自的均值和方差,可能会导致模型在不同 GPU 上产生不同的统计信息。SyncBatchNorm 可以在所有参与训练的 GPU 间同步计算批量归一化的均值和方差,确保所有 GPU 上的模型参数保持一致,从而提高模型的稳定性和收敛性。
if args.distributed and args.sync_bn:model = torch.nn.SyncBatchNorm.convert_sync_batchnorm(model)
5.优化器参数组:
这里使用了AdamW优化器。其中权重衰减是一种正则化技术,通过在损失函数中添加与模型参数的平方和成比例的惩罚项,防止模型过拟合。对于某些参数(如 bias 和 LayerNorm.weight),权重衰减可能是不必要的或不合适的,因此通常会对这些参数禁用权重衰减。
no_decay = ["bias", "LayerNorm.weight"]
optimizer_grouped_parameters = [{"params": [p for n, p in model.named_parameters() if not any(nd in n for nd in no_decay)],"weight_decay": args.weight_decay,},{"params": [p for n, p in model.named_parameters() if any(nd in n for nd in no_decay)],"weight_decay": 0.0,},
]optimizer = AdamW(optimizer_grouped_parameters, lr=args.lr)
三、下游应用
监督任务:细胞亚群的biomarker基因鉴定
Zero-shot任务:单细胞聚类降维效果比较、扩散拟时序分析、基因调控网络构建、

四、下游应用之监督任务
在预训练后的 iSEEEK 末端添加了一个线性分类器,并在 FACS 数据集分选的 CD4/8+ T 细胞上进行训练。把预训练好的iSEEEK参数冻结,对线性分类器进行参数更新。使用 Adam 优化器以 0.001 的学习率和 16 的批处理大小训练了这个分类器,训练了 30 个epoch。
任务本质是测量特定基因的影响,即原始基因序列的logit值与该基因序列被[UNK]标记替换的基因序列之间的差异。具体来说,对于S = [G1, G2, … , Gn]的原始输入基因序列,把G2替换为UNK得到了S∗ = [G1, UNK, … , Gn],来比较G2替换后对模型预测影响情况。
- logit 是 softmax函数的输入。例如,给定一个输入x,logit 值z是通过模型计算的,其中 W 是权重矩阵,b是偏置向量:

设 L 和 L∗ 分别表示从原本序列和替换序列获得的 logit 值。G2 对该分类器做出的决策的影响定义为:
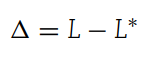
-
影响量 Δ代表的是模型预测强度(通过 logit 值衡量)的变化,度量的是这个特定基因在模型预测中的重要性。这代表了将某个基因替换为 [UNK] 标记后,对模型最终输出的预测分数的影响,可以用来判断哪些基因对于特定的细胞类型是关键的,从而识别出biomarker基因。
-
简单来讲,在没有替换之前的序列中,L值越大说明权重越大,表明该基因序列很重要。如果替换后L∗值也很大则说明这个基因换掉对模型的预测影响很小,换掉它无所谓,此时的就Δ很小。反之替换后L∗值很小,Δ则很大,说明换掉了比较重要的基因。
对于特定的细胞类型,通过细胞类型平均值对基因的影响进行排序,排名靠前的被认为是biomarker基因。
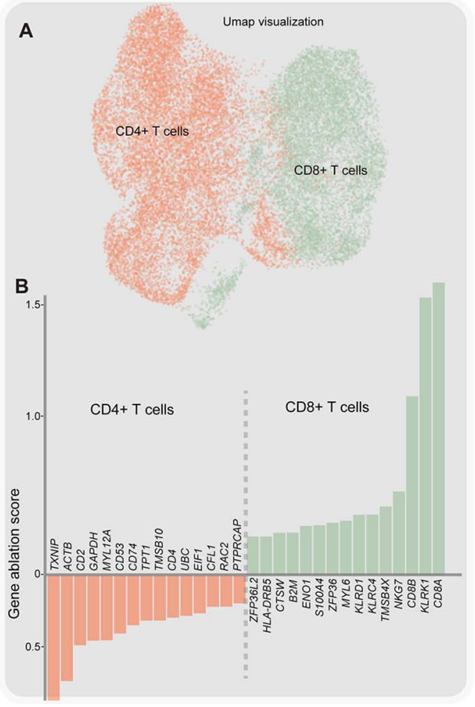
五、下游应用之Zero-shot任务–单细胞聚类降维效果比较
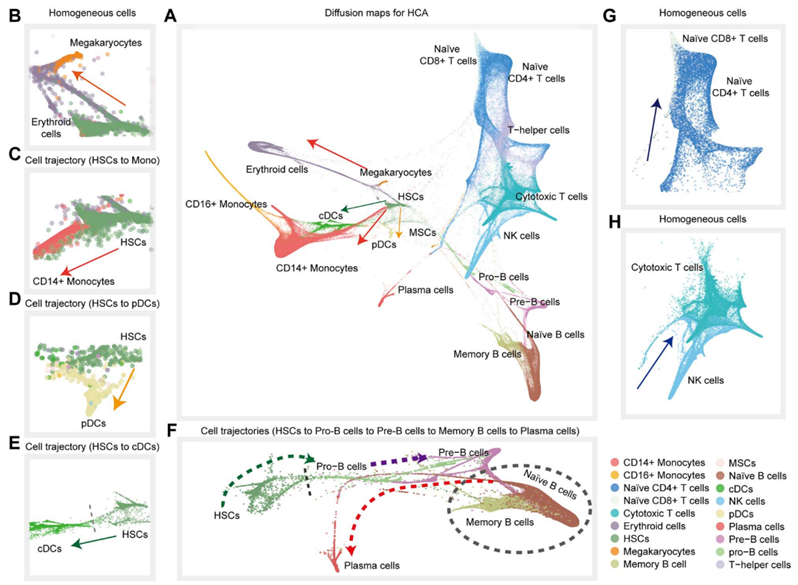
扩散拟时序分析(扩散图和扩散拟时序图是使用Pegasus包执行的,并使用强制定向布局嵌入算法可视化细胞轨迹。)
1.提取细胞表示特征
通过预训练的 iSEEEK 模型从单细胞数据中提取出的特征。这些特征本质上是模型对每个单细胞基因表达数据的高维表示。
def iseeek_feature(model, model_vocab, top_ranking_gene_list = []):Xs = []for s in tqdm(top_ranking_gene_list):a = ['[CLS]'] + s.split()[0:126] + ['[SEP]']input_ids = torch.tensor([model_vocab[k] for k in a]).unsqueeze(0).cuda()token_type_ids = torch.zeros_like(input_ids).cuda()attention_mask = torch.ones_like(input_ids).cuda()with torch.no_grad():feature = model(input_ids,token_type_ids,attention_mask)Xs.append(feature.cpu())Xs = torch.cat(Xs)features = pd.DataFrame(Xs.numpy(), columns=['Feature{}'.format(i) for i in range(Xs.shape[1])])return features
提取出来的特征如下(例子):
2.聚类降维
特征共有576个,对应隐藏层的维度。然后用提取出来的特征矩阵,跑scanpy的聚类降维流程,并与用基因表达矩阵跑聚类降维效果比较:

六、下游应用之Zero-shot任务–扩散拟时序分析
扩散拟时序分析(扩散图和扩散拟时序图是使用Pegasus包执行的。使用强制定向布局嵌入算法可视化细胞轨迹。)
1.亲和矩阵的构建
扩散拟时序分析(Diffusion Pseudotime, DPT)方法和预训练的基因BERT模型之间的联系在于输入特征的生成。预训练的基因BERT模型用于生成细胞的表示特征,这些特征随后作为输入用于扩散拟时序分析。
每个细胞的表示特征是通过基因BERT模型的 CLS token 提取的,其中CLS token 是整个输入序列(例如一组基因表达值)的全局表示,就是上面第五部分提取出来的特征矩阵。
利用特征矩阵计算得到亲和力矩阵(采用社区检测算法来构建,并用可导航小世界算法HNSW来查找top-k最近邻),其中的元素表示了细胞之间的相似性(或距离)。用缩放高斯核来代表细胞距离。
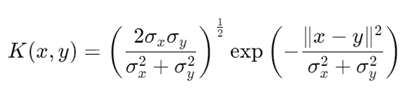
这里,x和y分别表示细胞x和细胞y的CLS token表示特征。 σ x σ_x σx 和 σ y σ_y σy 是局部核宽度,计算方式为该细胞与其最邻近k个细胞的中值。公式中的两部分分别描述了尺度因子(左)和高斯衰减项(右)。该距离度量的目标是结合局部核宽度来调整高斯核的平滑度,以捕获细胞间的相似性。
2.亲和矩阵的定义


这些相似性用于进一步的 Markov 链构建和扩散映射分析。
3.马尔可夫链转移矩阵和对称矩阵
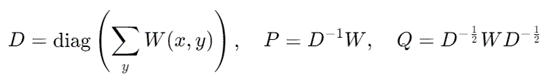
D 是对角矩阵,每个对角元素为所有与x相关联的W(x,y)的和;
P 是 Markov 过渡矩阵,表示在x和y之间的过渡概率,通过归一化 W得到;
Q 是对称矩阵,通过W进行两侧归一化得到。它用于进一步的谱分解,以构建扩散映射
4.Diffusion Maps
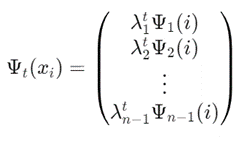
这是基于特征值 λ \lambda λ 的扩散映射。
每个 Ψ t ( x i ) \Psi_t(x_i) Ψt(xi) 是在时间尺度 t t t 上的扩散映射,其中 λ j \lambda_j λj 是 Q Q Q 的特征值, Ψ j ( i ) \Psi_j(i) Ψj(i) 是对应的特征向量。
这些扩散映射捕捉到了数据的流形结构,尤其是低维嵌入中的几何结构。
5.Diffusion Pseudotime Maps
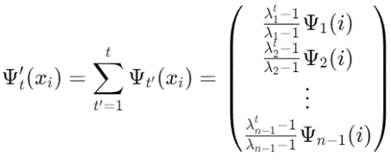
这表示扩散伪时间的映射。通过将不同时间尺度t′的扩散映射累加起来,可以捕捉数据中的时间演化特征。这些扩散伪时间映射用于推断细胞发展过程中的轨迹。
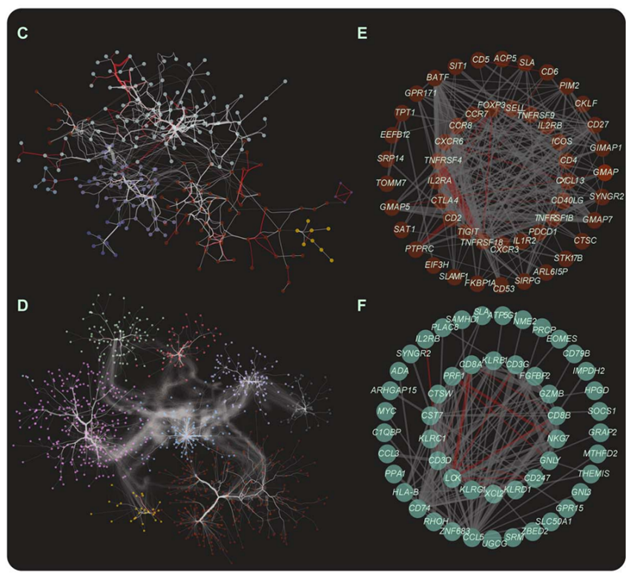
七、下游应用之Zero-shot任务–构建基因调控网络
目标是通过分析注意力矩阵来构建基因相互作用网络,并识别出具有统计学显著性的基因模块。
1.注意力矩阵α
注意力矩阵表示模型在进行决策时,如何在一组输入之间分配注意力。对于每对基因i和 j,注意力权重 a i , j a_{i,j} ai,j 表示模型在处理基因j时对基因i的关注程度。对于由n个基因组成的每个输入序列,可以提取一个n×n的注意力矩阵a,该矩阵中的每个元素 a i , j a_{i,j} ai,j 表示基因i 对基因j的注意力权重。
2.指标函数f(i,j,θ)
这是一个二值函数,用于过滤掉注意力权重较低的基因对。具体来说:如果基因i对基因j的注意力权重ai,j大于阈值θ,则 f(i,j,θ)=1,否则=0。
3.特定细胞类型的注意力矩阵 Cα(f)

C a ( f ) Ca(f) Ca(f) 是通过对细胞集合X中的所有细胞的注意力矩阵α累加得到的。这个矩阵综合了所有细胞的注意力信息,表示特定细胞类型中,基因对之间的整体注意力关系。
4.基因相互作用矩阵 G(i,j)
基因i和j之间的注意力 a i , j a_{i,j} ai,j与 a j , i a_{j,i} aj,i可能不同,矩阵 G(i,j)被定义为两者的加和
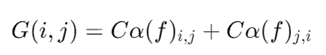
这个矩阵捕捉了基因对之间的双向相互作用,是对先前注意力矩阵的进一步细化和对称化处理。
5.网络构建与分析
通过保留G(i,j)中前10%的相互作用,构建一个稀疏的基因相互作用网络。这意味着只保留最强的相互作用,而忽略较弱的联系。
使用 Python 的 networkx 包来构建网络,并通过 Louvain 社区检测算法识别功能模块。
使用超几何检验来评估在 STRING 基因-基因相互作用数据库中检测到的模块的过表示情况,判断这些模块是否在生物学上具有显著性。
八、使用这个训练好的模型
1.准备好需要的包:
import pandas as pd
import os
import pickle
import torch
import numpy as np
from tqdm import tqdm
import matplotlib
import matplotlib.pyplot as plt
import matplotlib.colors as mcolors
import scanpy as sc
import pegasus as pg
import pegasusio
torch.set_num_threads(2)
2.iSEEEK FeatureExtractor:
def iseeek_feature(model, model_vocab, top_ranking_gene_list = []):Xs = []for s in tqdm(top_ranking_gene_list):a = ['[CLS]'] + s.split()[0:126] + ['[SEP]']input_ids = torch.tensor([model_vocab[k] for k in a]).unsqueeze(0).cuda()token_type_ids = torch.zeros_like(input_ids).cuda()attention_mask = torch.ones_like(input_ids).cuda()with torch.no_grad():feature = model(input_ids,token_type_ids,attention_mask)Xs.append(feature.cpu())Xs = torch.cat(Xs)features = pd.DataFrame(Xs.numpy(), columns=['Feature{}'.format(i) for i in range(Xs.shape[1])])return features
3.训练好的模型和Tokenizer载入:
提前下载好模型的权重和字典
!gdown https://drive.google.com/uc?id=1qorygy9HgJSGMgkv0QKdtDfW-K9o3wCY ### Download the File==Vocabulary of gene Tokenizer.
!gdown https://drive.google.com/uc?id=1WEc6v4mG1plPTPMaeLvl7hR1JGPHnUBn ### Download the File==Pre-trained iSEEEK Model.
model_vocab = pickle.load(open('iSEEEK_vocab.pkl',"rb")) ### Load the Vocalulary of gene Tokenizer.
genes = model_vocab.keys()
model = torch.jit.load("iSEEEK.pt") ### Load the iSEEEK model.
model = model.cuda()
model.eval()
print("###End loading model###")
4.数据准备:
提前下载好数据
!gdown https://drive.google.com/uc?id=1sLEMyCDv05nBGqHqFX6QoJ54RiguUrww
!gdown https://drive.google.com/uc?id=1RoP9ygs2oETIRif9royAzaB1CGlftK5m
!gdown https://drive.google.com/uc?id=1aLMDhZ6qtGsEJpDbazXEFhvq_0trQyx5
top_ranking_genes = [i for i in open("gene_rank_HCA_immune_processed.txt")]
label = [i for i in open("labels_HCA_immune_processed.txt")]
batch = [i for i in open("batch_HCA_immune_processed.txt")]
5.iSEEEK 特征提取:
iseeek_Xs = iseeek_feature(model,model_vocab,top_ranking_genes)
print(iseeek_Xs)
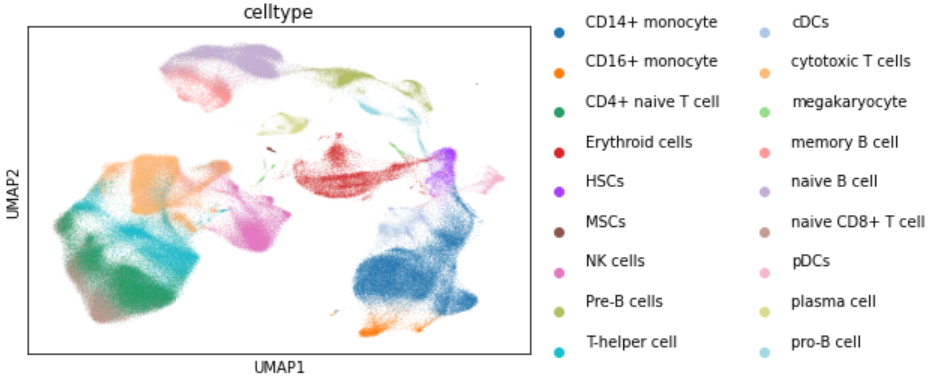
6.单细胞聚类降维:
adata = sc.AnnData(iseeek_Xs)
adata.obs['celltype'] = label
adata.obs['celltype'] = adata.obs['celltype'].astype("category")
adata.obs['batch'] = batch
adata.obs['batch'] = adata.obs['batch'].astype("category")
sc.pp.neighbors(adata, use_rep="X")
sc.tl.umap(adata)
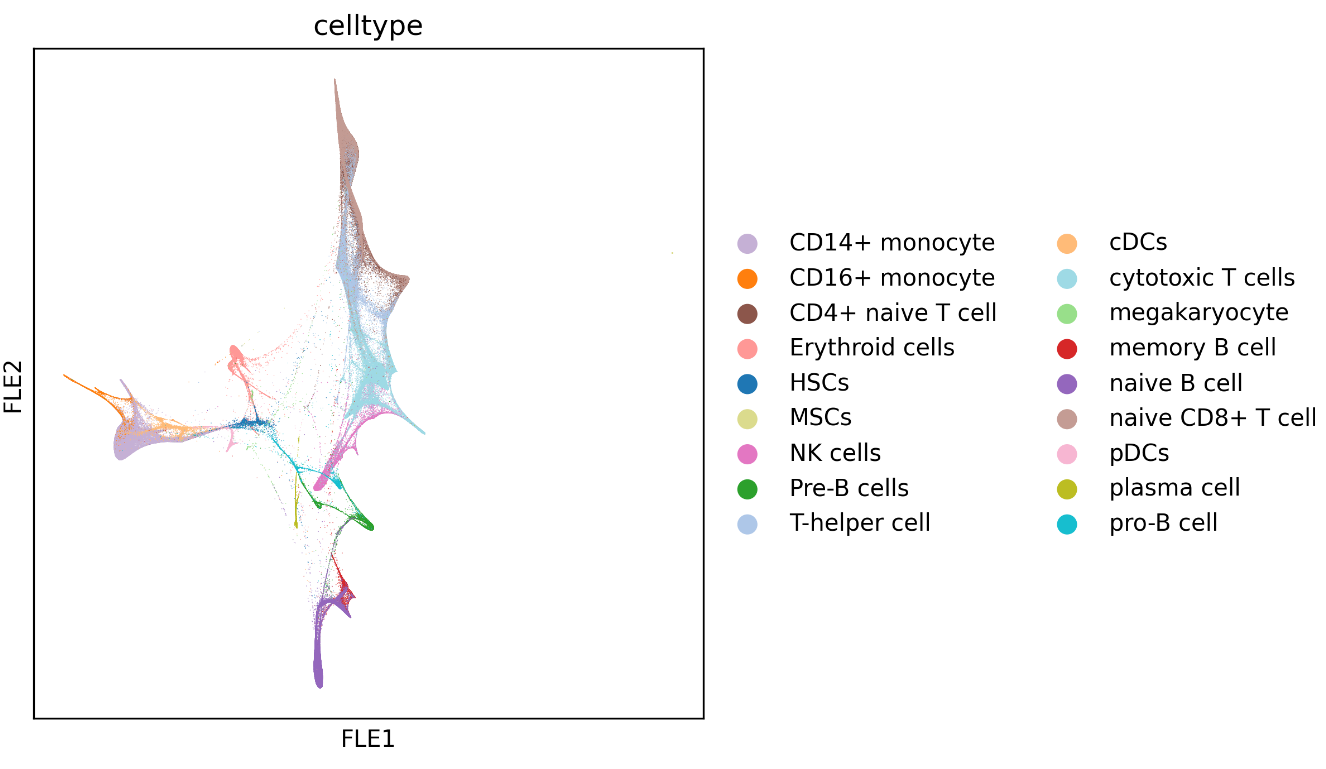
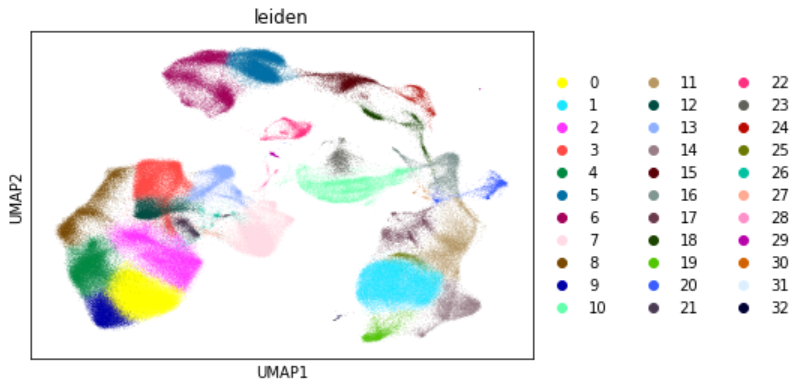
sc.tl.leiden(adata)sc.pl.umap(adata, color = ["celltype"], show = True)
sc.pl.umap(adata, color = ["batch"], show = True)
sc.pl.umap(adata, color = ["leiden"], show = True)

7.扩散拟时序分析:
adata = pegasusio.multimodal_data.MultimodalData(sc.AnnData(iseeek_Xs))
adata.obs['celltype'] = [i.strip() for i in label]
adata.obs['celltype'] = adata.obs['celltype'].astype("category")
adata.obsm["X_pca"] = np.asarray(iseeek_Xs)
pg.neighbors(adata,K =30)
pg.diffmap(adata)
pg.fle(data)pg.scatter(adata, attrs=["celltype"],show=True,basis='fle')