概述
论文地址:https://arxiv.org/abs/2401.04155
随着 OpenAI 的 GPT-X 和谷歌的 BERT 等大规模语言模型的出现,自然语言处理领域得到了飞速发展。这些先进的模型将理解和生成人类语言的能力发挥到了极致,彻底改变了日常交流和业务流程。
大规模语言模型通过研究互联网上的大量文本数据来学习语言的复杂性和语境,从而深刻理解文本的含义并做出适当的反应。这些模型的基础是一种名为 "转换器 "的创新型神经网络架构。这就实现了处理的并行化和可扩展性,同时捕捉文本的长程依赖关系。
特别值得一提的是转换器采用的 “自我关注机制”。在解释句子时,它会评估每个单词的重要性,从而加深对上下文的理解。这一技术是该模型取得卓越性能的关键。
学习分为两个阶段:预习和微调。在预学习阶段,使用大量的文本语料库来发展语法、事实知识和推理技能。通过微调,这些模型还能针对特定任务(如翻译、总结、问题解答)进行优化。它们的适应性使其能够处理各种自然语言处理任务,而无需依赖特定的架构。它们还具有应用于各种领域的潜力。
本文探讨了如何将大规模语言模型应用于各种问题。由于介绍的例子非常多,本文只选取其中一部分进行介绍。
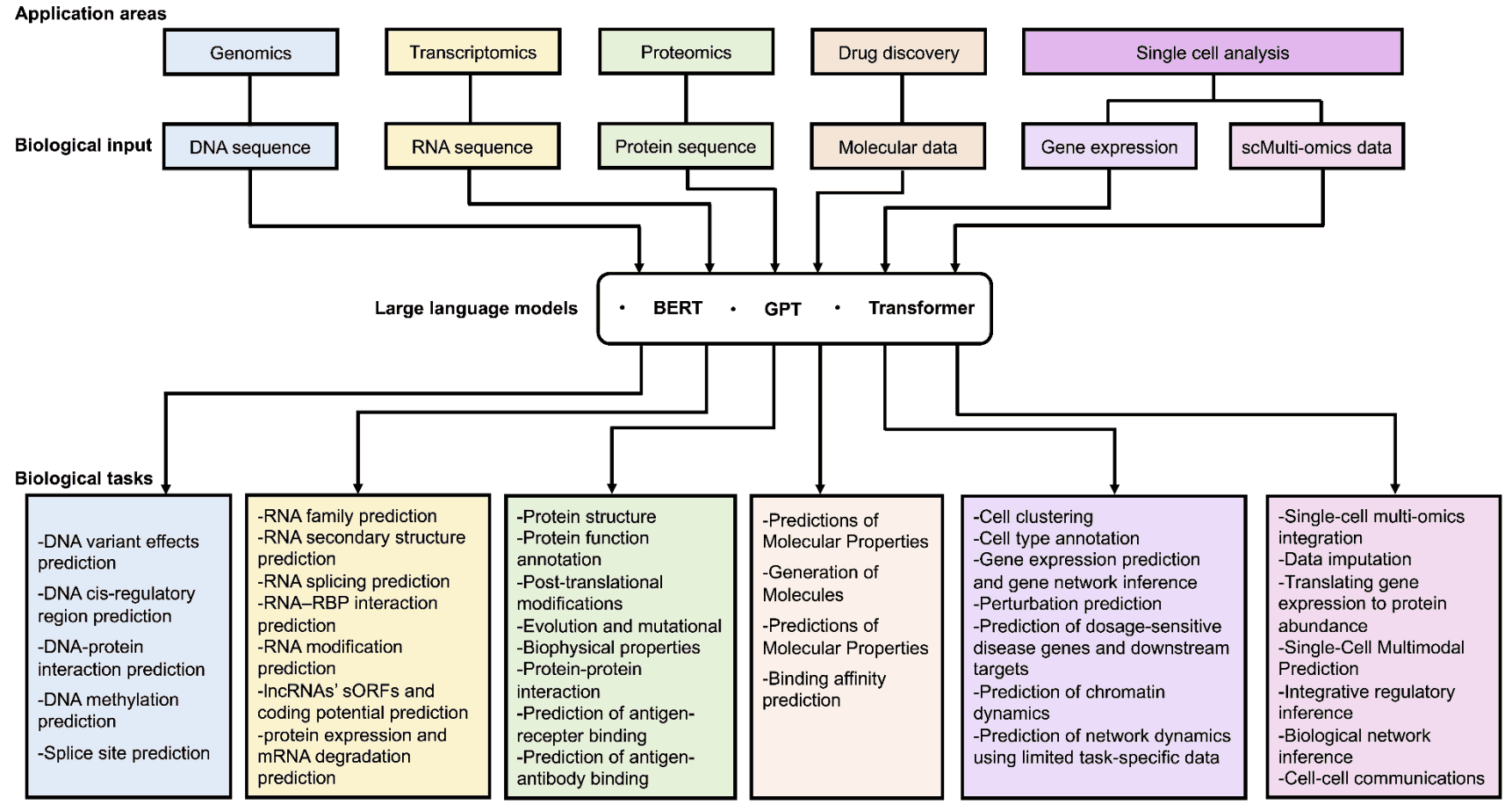
大规模语言模型在生物信息学中的应用
在生物学研究中,破译 DNA 中蕴含的语言并揭示隐藏的代码一直是一个主要目标。特别是通过使用 BERT 和 GPT 等现代架构的模型,在破译标志着 DNA 翻译成蛋白质的通用遗传密码方面取得了进展。
DNABERT 采用基于注意力的强大转换器架构,该架构已被广泛应用于各种自然语言处理任务。DNABERT-2 引入了基因组理解评估(GUE),这是一个用于多物种基因组分类的综合数据集。与之前的模型相比,该模型的效率提高了三倍,在使用的 28 个数据集中,有 23 个数据集的结果有所改善。
GROVER 还使用 DNA 语言模型,采用字节对标记化技术,对人类基因组进行详细分析。该模型可识别标记之间的上下文关系,并帮助识别与功能基因组注释相关的基因组区域结构。 GROVER 的独特方法对于探索基因组复杂性的研究人员来说非常宝贵。
此外,DNAGPT 是在 GPT 系列取得成功后开发的,它是基于 GPT 的 DNA 模型,已在超过 100 亿个碱基对的数据集上进行了预训练,可针对各种 DNA 测序分析任务进行微调。核苷酸转换器还开发了四种不同规模的语言模型,并在涵盖多个物种的三个不同数据集上进行了预训练。
这些预训练模型已被应用于多种序列预测任务,包括启动子区域、增强子区域、顺式调节元件、剪接位点和转录因子结合位点的预测。
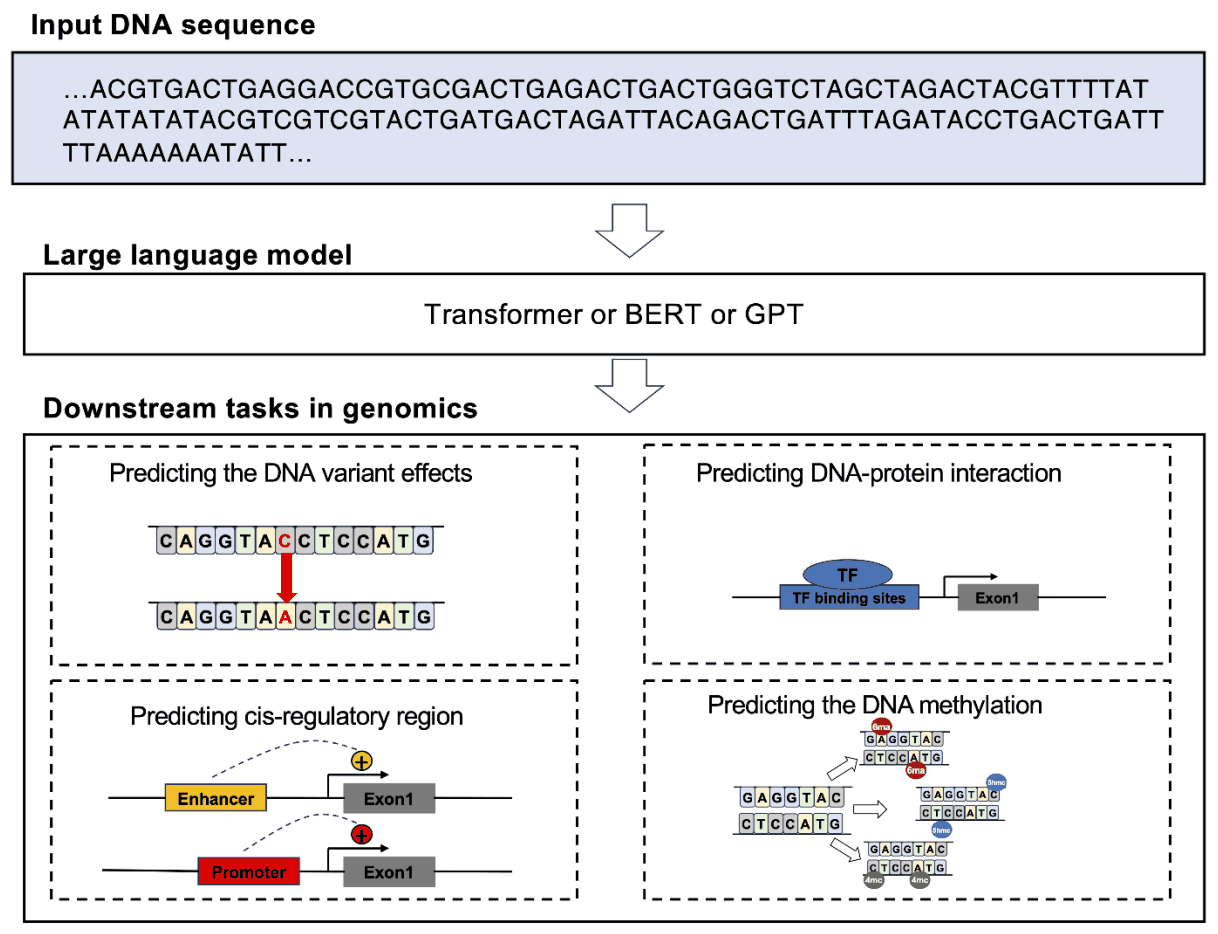
大规模语言模型在生物信息学中的一个应用是 DNA 序列语言模型预测全基因组突变效应DNA 变异对生物多样性的重要性是巨大的。全基因组关联研究(GWAS)在阐明这一点方面发挥着重要作用,但识别因果变异却是一项重大挑战。为应对这一挑战而开发的基因组预学习网络(GPNs),旨在通过无监督预学习获得全基因组变异效应的知识;GPNs 使用特定位置屏蔽的 512 碱基对 DNA 序列预测核苷酸,然后可用于识别全基因组的变异效应、它特别擅长准确捕捉罕见突变的影响。它已证明有能力从一系列物种的 DNA 序列中预测突变效应,而且该技术有助于正在进行的研究,以了解 DNA 序列突变与生物多样性之间的复杂关系。
第二个是预测顺式调控区域的 DNA 序列语言模型。鉴别调控基因表达的顺式调控序列(尤其是增强子和启动子)至关重要,因为它们对发育和生理功能都有影响。识别这些序列是一项重大挑战,而 DNABERT 和 GROVER 等预训练模型的开发就是为了提高识别的准确性。例如,BERT-Promoter 使用预先训练的 BERT 模型来识别启动子活性,并应用先进的机器学习算法来建立最终的预测模型。另一方面,iEnhancer-BERT 使用基于 DNABERT 的过渡学习方法来增强启动子预测,并使用卷积神经网络对特征向量进行分类。这些模型在揭示基因表达背后的机制和识别新的 DNA 增强子方面取得了可喜的进展。
第三是预测 DNA 蛋白相互作用:准确识别 DNA 蛋白相互作用对于理解基因表达调控和进化过程至关重要。DNABERT、DNABERT-2 和 GROVER 等模型就是针对这一重要任务开发的,它们根据 ChIP-seq 数据预测蛋白质与 DNA 的结合。TFBERT 也是一种预先训练好的模型,只需极少的微调就能表现出色。该模型像对待自然语言一样对待 DNA,能有效提取上下文信息,从而高效地完成任务。另一方面,MoDNA 框架纳入了常见的 DNA 功能主题,并通过自监督预训练获得基因组表征,有助于启动子预测和转录因子结合位点预测。
第四个是DNA 甲基化预测,这一过程在基因的表观遗传调控中发挥着核心作用。甲基化模式是诊断和治疗疾病的重要标志。有几种进化模型,特别是 BERT6mA、iDNA-ABT、iDNA-ABF 和 MuLan-Methyl,可以预测不同形式的甲基化,这些洞察力可以促进新疗法的开发。这些模型利用先进的特征表示和学习算法来分析 DNA 甲基化的复杂模式。
大规模语言模型在转录组中的应用
随着基于 BERT 的 DNA 语言模型的不断发展,如何从异源序列中准确捕捉进化信息成为了一项挑战。特别是出现了两种基于 RNA 的创新模型,即 RNA-FM 和 RNA-MSM,以处理保守程度较低的 RNA 序列。
RNA-FM 利用自我监督学习,使用包含 2,300 万个非编码 RNA 序列的广泛数据集预测 RNA 二级和三维结构。通过这种方法,RNA-FM 能有效捕捉 RNA 序列的各种结构信息,全面了解这些序列的特征。
另一方面,RNA-MSM 利用 RNAcmaps 自动收集的同源序列。该模型尤其擅长精确绘制二维碱基配对概率和一维溶剂可及性,可对与 RNA 结构和功能相关的各种下游任务进行微调。
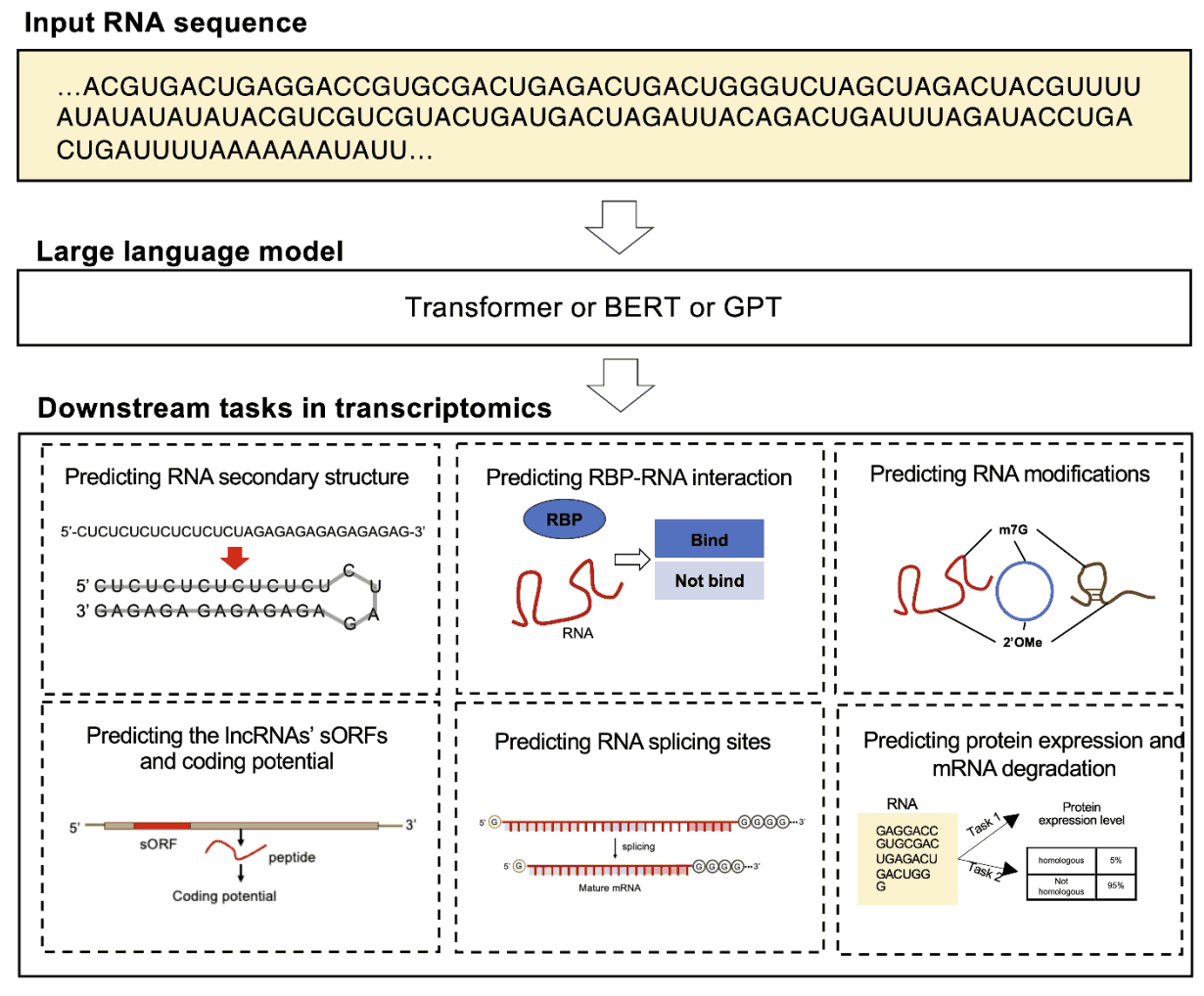
大规模语言模型在转录组中的一个应用是利用 RNA 序列语言模型预测 RNA 家族分类和二级结构。RNA 二级结构预测是科学家深入了解 RNA 折叠规则所面临的一大挑战。RNABERT 就是为应对这一挑战而开发的,它有可能为包括开发 RNA 靶向药物在内的许多应用做出贡献。它结合了标记化、位置嵌入和转换器建模,尤其侧重于预测 RNA 二级结构和 RNA 家族分类。这种理解复杂 RNA 折叠规则并将未知 RNA 序列快速归入现有 RNA 家族的能力,是研究新 RNA 分子的重要进步。RNABERT 是提高我们对整个 RNA 生物学以及 RNA 结构预测的认识的重要工具。
第二个是用于预测RNA 剪接的 RNA序列语言模型,RNA 剪接是真核生物基因表达的重要过程。为了更好地理解这一过程,我们开发了一个名为 SpliceBERT 的预训练模型。SpliceBERT 不仅能捕捉 RNA 剪接的细微差别,还能帮助识别阻止剪接的潜在突变。它有助于识别阻止剪接的潜在突变。该模型提供了一种数据驱动的方法,有助于评估突变的影响,并有效地识别重要的基因变异,确定其优先次序。这种能力是研究人员了解 RNA 剪接影响的宝贵资源。
第三种是用于预测 RNA 修饰的 RNA 序列语言模型:RNA 的转录后修饰在细胞中发挥着重要作用,尤其是 N7-甲基鸟苷(m7G)等修饰,对基因表达的调控至关重要。高通量实验虽然准确,但成本高、耗时长。为此,BERT-m7G 应运而生,它是一种从 RNA 序列中有效识别 m7G 位点的计算模型。这一工具减轻了实验方法的负担,有助于更好地了解 m7G 如何影响基因功能。此外,另一种 RNA 修饰–2’-O-甲基化(Nm)对细胞过程也很重要,Bert2Ome 是一种高效的计算工具,可直接从 RNA 序列中预测该位点。 基于 bERT 的模型与卷积神经网络(CNN)相结合将基于 BERT 的模型与卷积神经网络 (CNN) 结合使用,可以高精度地识别 RNA 修饰位点及其功能关系。这种方法大大缩短了实验方法所需的时间,有助于人们对转录后修饰有新的认识。
第四个是用于预测蛋白质表达和 mRNA 降解的 RNA 序列语言模型。mRNA 疫苗因其成本效益和快速发展潜力而备受关注。codonBERT 是专门设计用来预测 mRNA 序列中的蛋白质表达的,它是一种使用头部注意力转换器架构在广泛的数据集上进行预训练的多元智能。这种预训练使 CodonBERT 在预测蛋白质表达和 mRNA 降解方面具有卓越的性能,并能将新的生物信息纳入 mRNA 疫苗的设计中。该模型为免疫领域带来了新的可能性,有助于更高效地开发疫苗。

大规模语言模型在蛋白质研究中的应用
蛋白质是维持生命的重要分子,是各种生理过程的基础。随着科学的发展,蛋白质数据也在不断积累。大规模语言模型正在成为从这些数据中提取有用信息的有效手段。
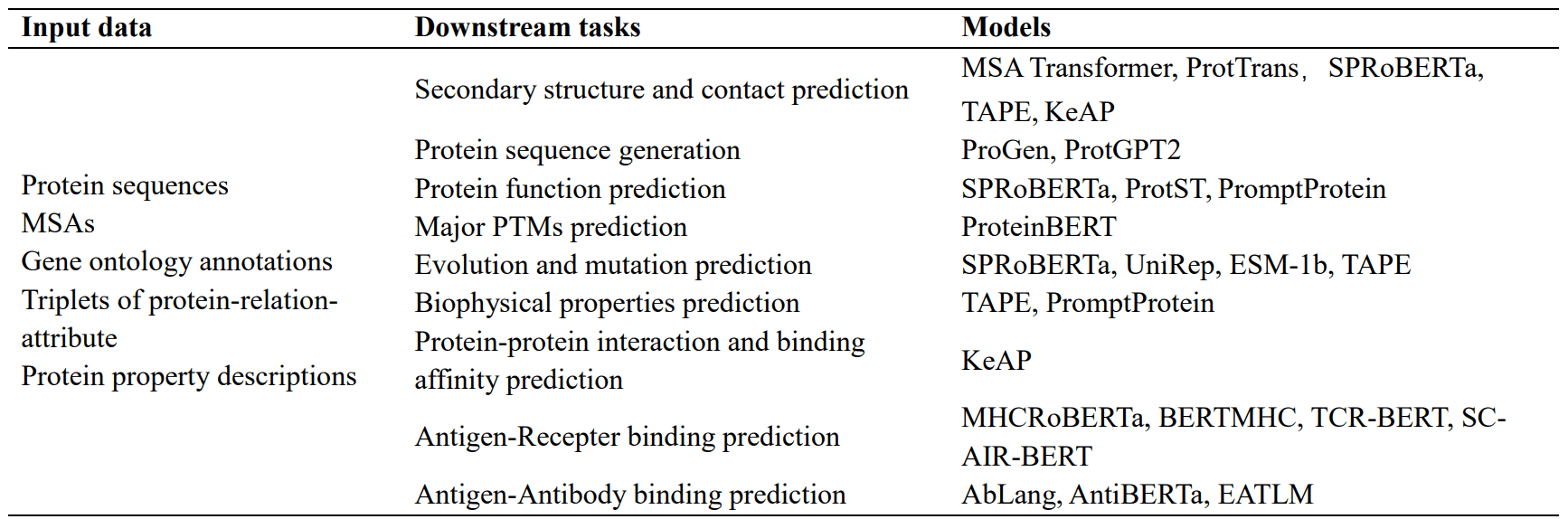
这些模型被称为预训练蛋白质语言模型(PPLM),可从蛋白质序列、基因本体注释和属性描述等数据中学习特征。学习到的特征可用于各种任务,如蛋白质结构预测、翻译后修饰(PTM)和生物物理特性评估。
抗体也是蛋白质的一种,但用于研究抗体的数据集和任务与一般蛋白质不同。观察到的抗体空间(OAS)数据库不断扩大,促使人们开发出了抗体特异性大规模语言模型(PALM),用于研究治疗性抗体的结合机制、免疫进化和新抗体的发现。这些模型可用于预测特定的抗体位点(旁位点)、分析 B 细胞的成熟过程以及对抗体序列进行分类等多种任务。
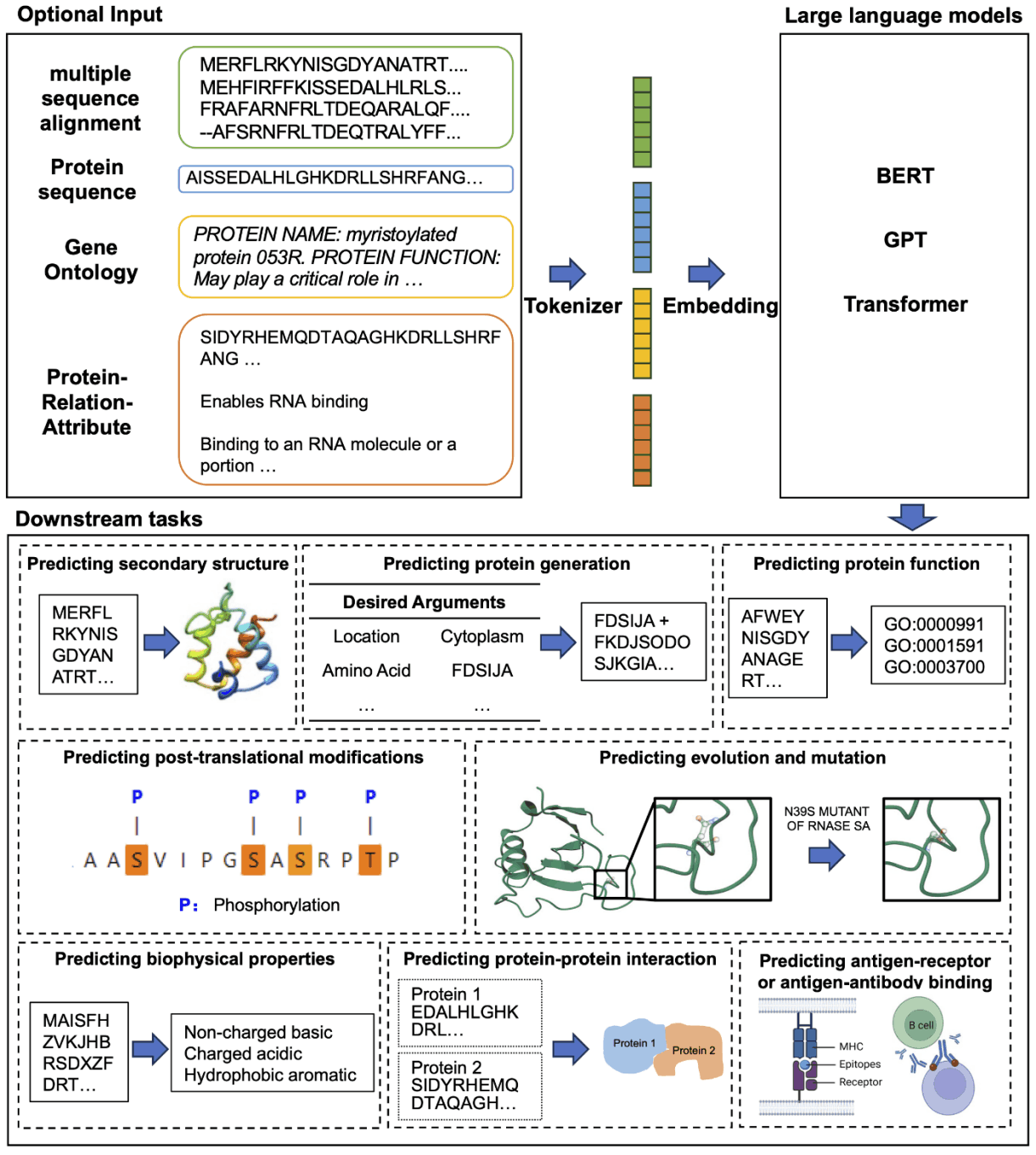
大规模语言建模在蛋白质研究中的一个应用是蛋白质二级结构和接触前理解的蛋白质语言建模。蛋白质结构对其功能和相互作用起着至关重要的作用。然而,利用传统实验室技术分析蛋白质结构是一个耗时耗力的过程。为解决这一问题,深度学习技术的进步催生了用于预测蛋白质结构的大规模语言模型。例如,MSA Transformer 是一种使用多序列比对并对输入序列应用独特的行列关注机制的模型。该模型优于传统的无监督方法,并提高了参数效率。ProtTrans 还使用 UniRef 和 BFD 的数据来训练多个模型,在预测二级结构方面取得了重大进展。
第二个是用于生成蛋白质序列的蛋白质语言模型。蛋白质生成技术应用广泛,包括药物设计和蛋白质工程。现代大规模语言模型有能力在生成蛋白质序列时形成具有特定功能特性的稳定三维结构,ProGen 模型使用 UniprotKB 关键词作为条件标签,在超过 1,100 个术语的丰富词汇中进行它生成。此外,ProtGPT2 生成的蛋白质遵循自然界的氨基酸原理,其中许多蛋白质具有球状特性,而当使用 AlphaFold 时,数据库中以前不存在的新拓扑结构也被揭示出来,这表明 ProtGPT2 正在学习一种蛋白质专用语言并获得了
第三个是用于预测抗原-受体结合和抗原-抗体结合的抗体大规模语言模型。抗原蛋白在细胞质中降解形成新的抗原肽。这些肽与主要组织相容性复合物(MHC)结合,形成 pMHC 复合物,并被转运到细胞膜上,在细胞膜上呈现;T 细胞受体(TCR)识别这些肽,并刺激 B 细胞产生特异性抗体,从而引发免疫反应。这一过程的一个重要部分是预测多肽与 HLA 分子结合的准确性。
例如,MHCRoBERTa 可以根据输入的氨基酸序列区分不同的等位基因,但该模型专门用于 pMHC-I 结合预测。另一方面,BERTMHC 已在包含 2,413 对 MHC 肽对的数据上进行了训练,并在填补 pMHC-II 结合预测的空白方面取得了进展。
另一个主要目标是预测适应性免疫受体(AIR)与抗原结合的特异性。这种特异性主要归因于三个互补性决定区(称为 CDR1-3)环的灵活性,TCR-BERT 从未加标记的 TCR CDR3 序列中学习 TCR 的一般表示,从而预测抗原的特异性。然而,这一模型在理解 AIR 对的相互作用方面并不成功。姚建华等人专门设计的BERT模型SC-AIR-BERT有效地解决了这一问题,该模型在预测TCR和BCR的抗原结合特异性方面优于其他方法。
最近关于抗体语言模型的研究也引起了人们的关注。例如,AbLang 建立在 RoBERTa 的基础上,专注于特定的挑战,特别是恢复测序过程中丢失的残基。该模型在准确恢复抗体序列中丢失的残基方面优于其他模型。
此外,AntiBERTa 还利用源自蛋白质序列的潜在载体来了解抗体的 “语言”,并通过引入额外的预学习任务来有效地执行各种任务,如跟踪抗体的 B 细胞来源、量化免疫原性和预测结合位点 EATLM、EATLM 通过引入额外的预学习任务、新的方法,将特定的生物机制纳入其中。
大规模语言模型在药物发现中的应用
众所周知,药物发现是一个成功率低、成本高、耗时长的过程。在这一早期阶段,计算机辅助药物发现将算法、机器学习和深度学习与经验和专业知识相结合,正在加速药物分子和先导化合物的生成和筛选。这加快了整个开发过程,尤其是小分子药物的开发,而小分子药物往往占市场上药物的大部分(高达 98%)。
小分子药物因其结构具有良好的空间分散性和化学性质,被认为具有良好的类药物和药代动力学特性。深度学习的进步和大规模语言模型的引入,使得利用这些技术发现小分子、蛋白质、RNA 和其他靶标之间的模式和相互作用变得更加容易。
具体来说,SMILES 字符串和化学指纹常用于表示分子。此外,图神经网络(GNN)的汇集过程可用于将小分子转换为序列表示,大规模语言模型则可在这些信息的基础上用于药物发现的不同方面。这样,新药发现的效率和准确性就得到了提高。
这种方法为降低药物发现领域的成本和加快进程做出了重大贡献,为未来的医疗保健开辟了新的可能性。
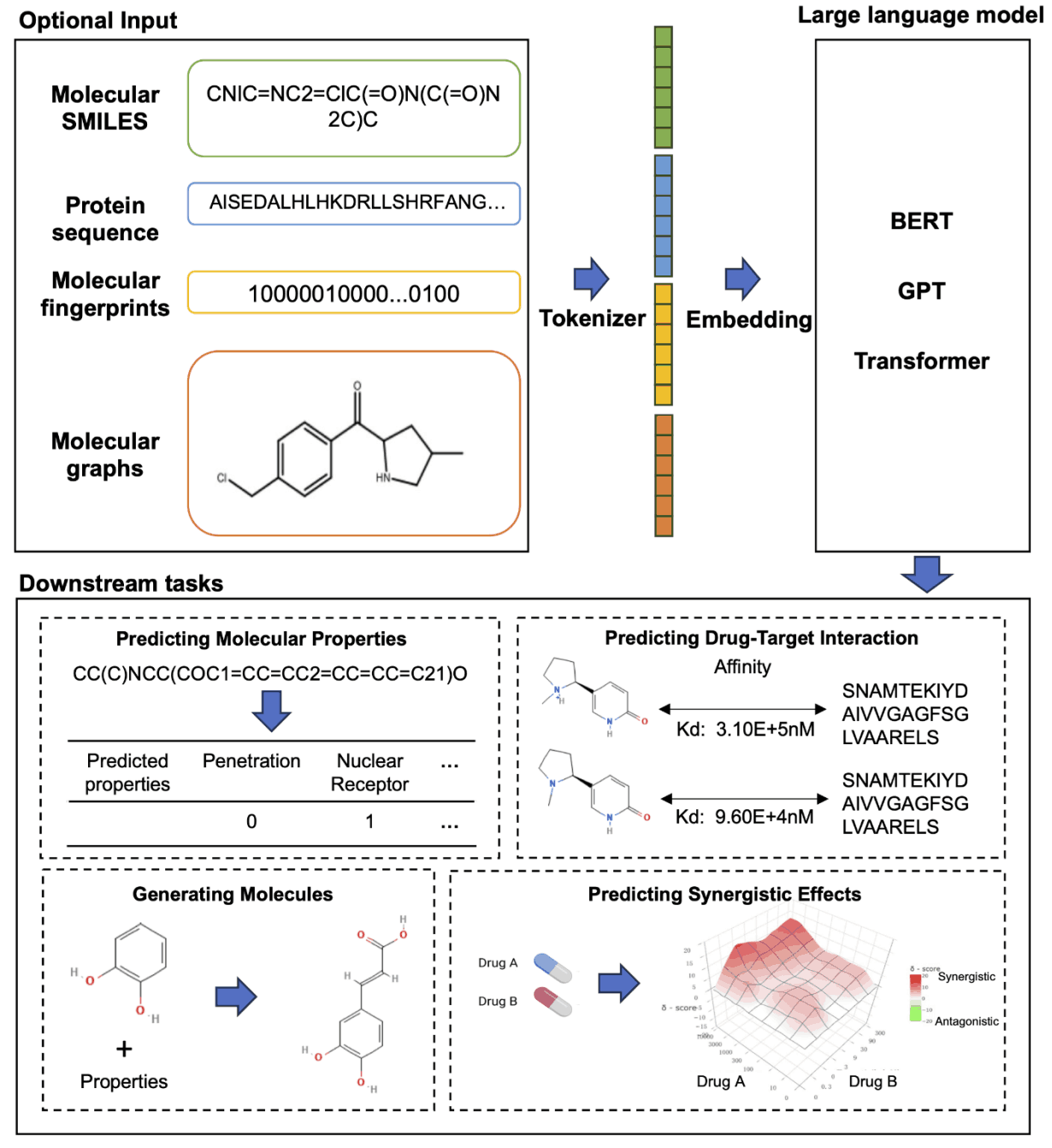
大规模语言模型在药物发现中的一个应用是,覆盖大量的类药物化学空间(估计超过 10 至 63 次方的化合物)是一项非常艰巨的实际挑战。传统的虚拟筛选库仅包含少于 10 至 7 次幂的化合物,有时甚至无法获得。为解决这一问题,深度学习方法已成为生成具有类药物特性的分子的有效方法。其中,MolGPT 模型受预生成学习模型 GPT 的启发,通过纳入下一个标记预测任务以及条件预测的额外训练任务,扩展了条件生成的能力。该模型不仅能生成创新而有效的分子,还能增强理解数据集统计特性的能力。
大规模语言建模在药物发现中的第二个应用是,治疗癌症、传染病和神经系统疾病等复杂疾病的联合疗法很常见,而且往往比单一药物治疗更有效。准确预测药物组合的协同效应对提高疗效至关重要,但由于药物组合数量庞大,生物相互作用复杂,因此具有挑战性。在这一领域,张伟及其同事开发的 DCE-DForest 模型使用预先训练的药物 BERT 模型对药物 SMILES 进行编码,并使用深度森林方法从药物和细胞系的嵌入向量中预测协同效应。此外,Mengdie Xua 等人对预训练的大规模语言模型进行了微调,并使用双特征融合机制有效地预测了药物配对的协同作用。这包括药物分子指纹、SMILES 编码和细胞系基因表达数据,去除分析证实指纹输入对药物协同作用预测的质量起着重要作用。

大规模语言模型在单细胞分析中的应用
单细胞 RNA 测序(scRNA-seq)标志着基因组学和生物医学研究新时代的开始。与传统的批量 RNA 测序不同,scRNA-seq 可以在单细胞水平上揭示基因表达的细节,从而带来前所未有的洞察力和许多突破 [127-130]。这项技术带来的最显著的变化之一是能够详细了解组织或生物体内细胞的多样性。传统方法往往忽略的多种细胞类型和罕见细胞状态,都可以通过 scRNA-seq 得到揭示。
如上所述,大规模语言模型已成功应用于基因组学、转录组学、蛋白质组学和药物发现等多个领域。在此,我们将展示这些模型是如何应用于单细胞分析领域的。单细胞语言模型可用于多种下游任务,如识别细胞类型和状态、发现新细胞群、估计基因调控网络,甚至整合单细胞多组学数据。
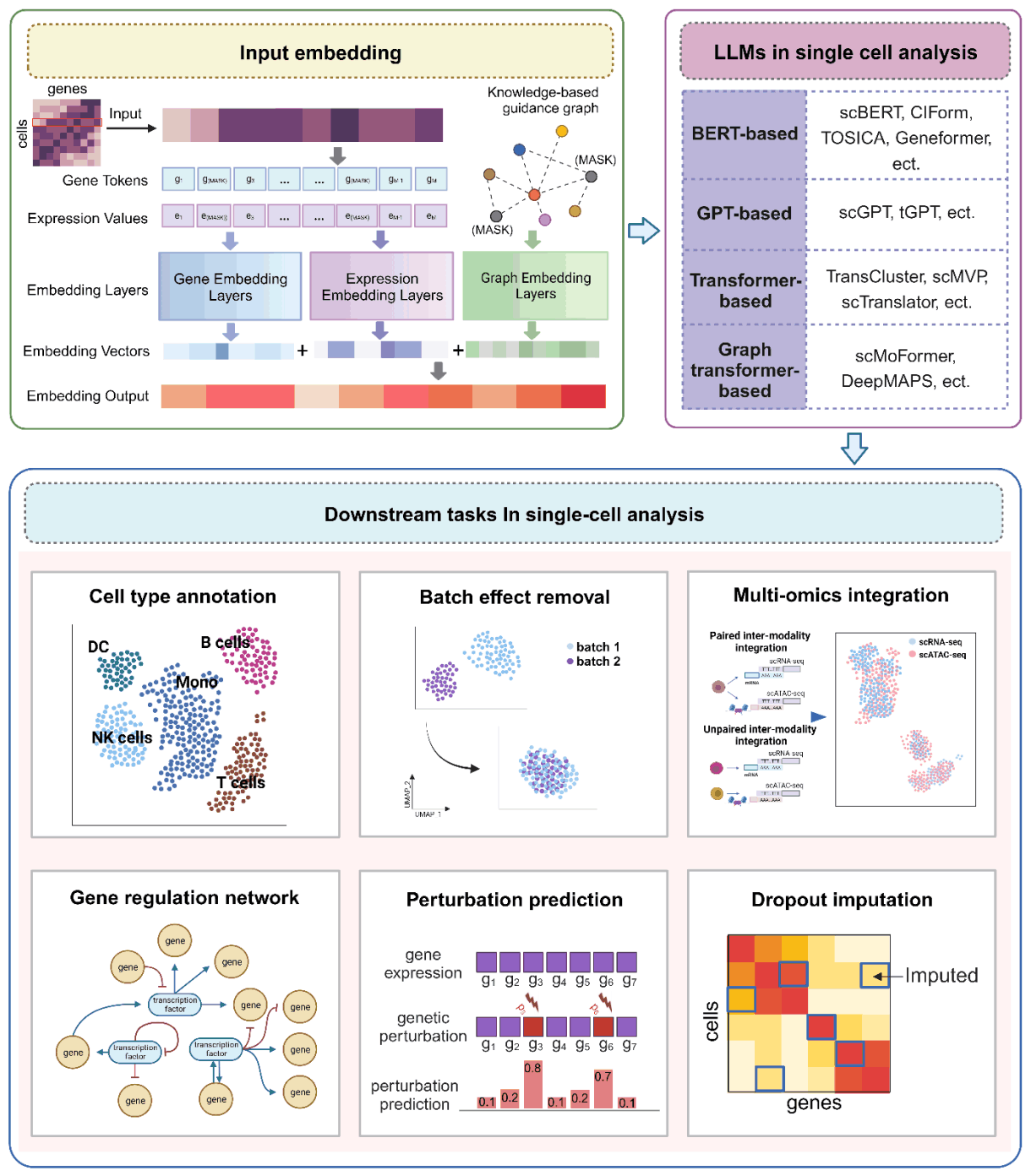
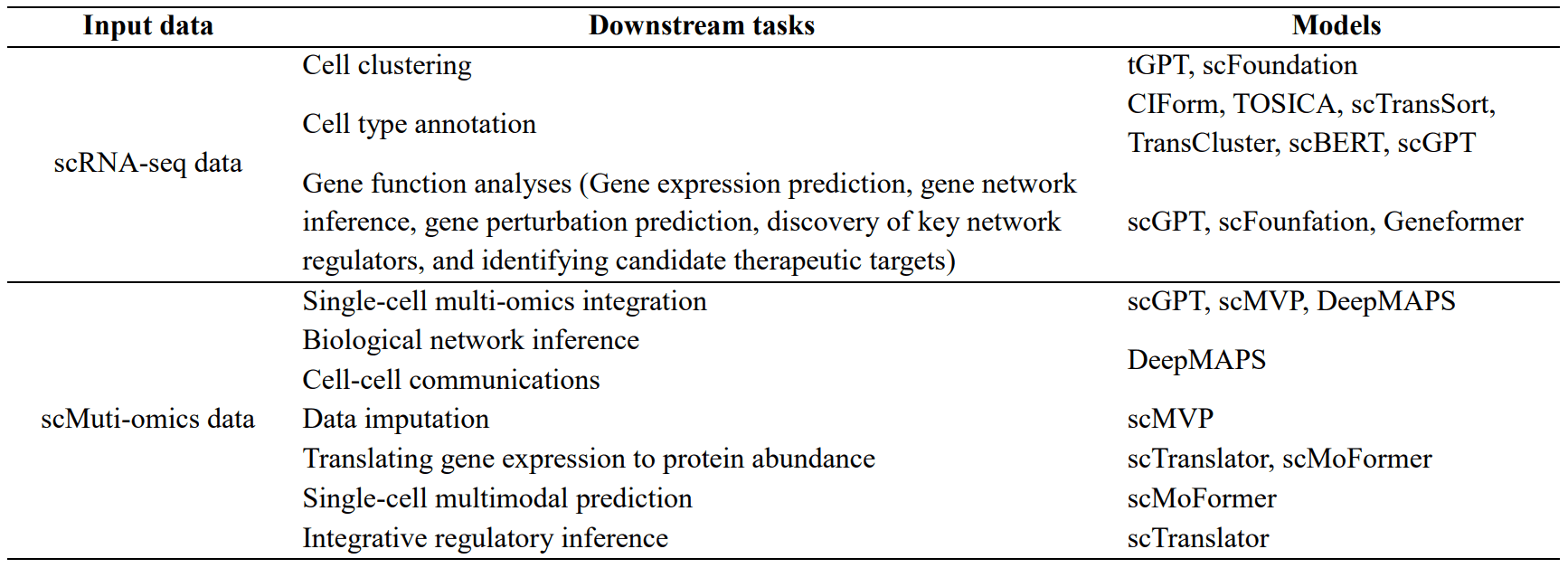
大规模语言模型在单细胞分析中的一个应用是基于 scRNA-seq 数据进行单细胞聚类的单细胞语言模型。通过单细胞 RNA 测序(scRNA-seq)进行细胞聚类是解读生物样本中细胞多样性的重要方法。它可以根据基因表达谱将单个细胞划分成群。大规模语言模型可以利用来自不同组织和物种的大量 scRNA-seq 数据进行高效聚类。例如,tGPT 模型可根据高表达基因学习特征表征,并已应用于人类细胞图谱和 Tabula Muris 等大型数据集的细胞聚类。scFoundation 还使用基于变换器的编码器-解码器结构,从未加掩码的非零基因数据中学习细胞嵌入,然后用于聚类。
第二个是基于 scRNA-seq 数据的基因功能分析单细胞语言模型。大规模语言模型也被应用于基因功能分析。这些模型利用转化注意机制学习基因之间的关系,并通过预训练和微调生成基因嵌入。这些嵌入可用于基因表达预测和遗传扰动预测。scGPT 利用零点学习作为特征提取器,有助于推断基因调控网络。另一方面,Geneformer 在大量单细胞转录组数据的基础上进行训练,并针对各种下游任务(如预测染色质动力学和网络动力学)进行微调。这些模型通过将预先训练好的权重转移到特定任务模型中,在数据有限的情况下提供高精度的预测。
第三是单细胞多组学数据的单细胞语言模型。与单细胞组学数据类型相比,单细胞多组学数据的研究通过在单细胞水平上整合基因组学、转录组、表观基因组和蛋白质组等不同组学技术的信息,具有许多优势。在分析这类数据时,大规模语言模型凭借其适应性、通用性和特征提取能力,为解决数据多变性、稀缺性和细胞异质性等难题提供了解决方案。
在整合 scMulti-omics 数据时,一个名为 scGPT 的模型通过使用代表不同测序方法的标记集来处理数据集的多样性。这些模式标记与基因或蛋白质等输入特征相关联,并纳入转换器输出,以提高数据处理的准确性。这种独创性可避免过度关注同一模式中的特征,同时允许对不同模式中的特征进行适当评估。
特别值得注意的是 scMVP 工具,它是专门为整合单细胞 RNA-seq 和 ATAC-seq 数据而设计的,在同一细胞中分析基因表达和染色质可及性。类型引导的注意函数来计算数据之间的相关性。DeepMAPS 则是一种基于嫁接变换器的方法,用于从 scMultiomics 数据(包括 scRNA-seq、scATAC-seq 和 CITE-seq)中进行生物网络推断和数据整合。该方法以基因和细胞为节点构建图,并学习区域和全局特征以建立细胞和基因之间的关系。
scTranslator 还能将单细胞转录组数据转换为蛋白质组数据,并通过最大限度地减少预测蛋白质与实际蛋白质之间的差异来准确推断蛋白质丰度。scMoFormer 不仅能将基因表达转换为蛋白质丰度,还能预测多组学数据,揭示不同生物信息之间的动态交互。
因此,大规模语言模型在单细胞分析领域发挥着重要作用,开辟了新的研究可能性。这些不断发展的工具为揭示生物复杂性提供了新的可能性,并为精准医疗铺平了道路。
总结
预训练的大规模语言模型正在彻底改变生物学中的各种挑战。本文探讨了大规模语言模型在基因组学、转录组学、蛋白质组学、单细胞分析和药物发现等多个领域的应用。
大规模语言模型分析 DNA 和 RNA 序列,在此基础上预测修饰和调控。蛋白质组学领域也取得了重大进展,包括预测蛋白质结构和相互作用。特别是,来自 scRNA-seq 和 scMulti-omics 数据的信息有助于确定细胞类型、整合数据集和预测与基因有关的功能分析。
在药物发现方面,大规模语言模型也被用于预测分子特性、预测新分子的生成和药物相互作用。例如,DNABERT 是专门为 DNA 分析而训练的,也可应用于 RNA 分析;M6A-BERT-Stacking 等模型专门用于识别 RNA 修饰位点,可以做出高度准确的预测。
在蛋白质研究领域,基于测序数据的蛋白质语言模型可对蛋白质功能进行详细分析,并为研究人员提供有用信息。然而,这些模型需要大量参数,这给部署带来了挑战。部分解决方案是利用大规模在线模型或基于蒸馏的方法。
因此,大规模语言建模作为分析分子生物学复杂问题(从分析 DNA 突变和 mRNA 丰度到发现新的因果关系)的有力工具,正在开辟新的可能性。
此外,大规模语言模型的开发为整合蛋白质三维结构信息等不同信息模式带来了新的挑战。目前正在研究将这些信息转换为基于序列的格式的方法,以及整合多个大型模型以捕捉多模态信息的方法。多模态融合技术和时机选择对此非常重要。
在药物发现领域,大规模语言模型的使用不仅需要基于分子序列信息的预测,还需要基于分子空间结构的预测。例如,利用 CrossDocked2020 数据集开发了大规模图形模型。
此外,大规模语言模型可将从预测蛋白质-蛋白质相互作用(PPI)和细胞-细胞相互作用(CCI)中获得的知识应用于预测药物-靶点相互作用(DTI)。考虑到药物的疗效和新颖性等特性,这项技术在生成药物分子方面也在不断发展。
大规模语言模型在单细胞分析中的应用减少了稀缺性问题,尤其是 scRNA-seq 数据,并简化了基于大量基因表达数据的模型训练。在整合来自不同测序技术的数据时,定义基因位置信息和克服批次效应也是重要的挑战。图神经网络(GNN)与转换器的结合正在推动单细胞分析的创新进展,有助于分析复杂的细胞-基因相互作用。
DeepMAPS 还是一个使用嫁接变换器评估细胞内基因重要性的模型,以了解细胞与基因之间的相互作用。该技术综合利用图神经网络(GNN)和转换器来全面表示单细胞数据中存在的复杂关系和依赖性;GNN 适合捕捉细胞附近的局部相互作用,而转换器则能更有效地捕捉更广泛的依赖性。而转换器则能更有效地捕捉更广泛的依赖关系。
这种协同作用有助于了解细胞的整体情况并改进特征学习。因此,大规模语言模型可以有效地从原始数据中学习基因表达模式和细胞类型等相关信息,而无需事先了解特定领域的知识。
当今的大规模语言模型在模拟复杂的分子生物学方面已经达到了非常复杂的水平。单细胞技术的发展以及包括蛋白质组学、代谢组学和脂质组学在内的全方位科学(omics science)的发展,使得更高效的测量技术成为可能。这增强了我们揭示从 DNA 到人体生理细节等分子层复杂性的能力。
对这一尖端技术领域的进一步探索有望为全面了解分子水平的动态相互作用提供新的视角。