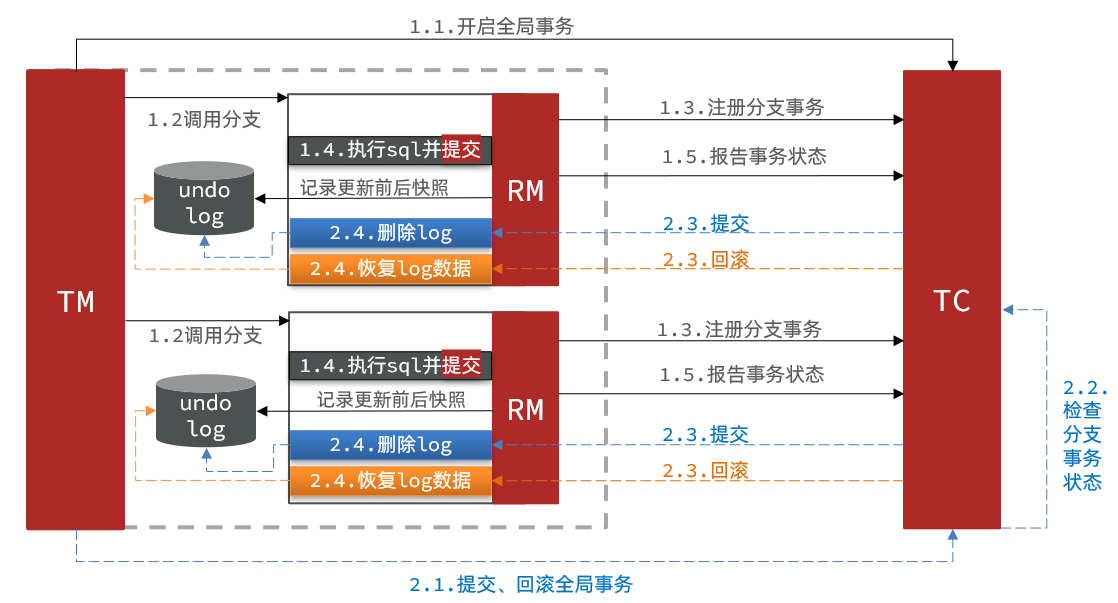
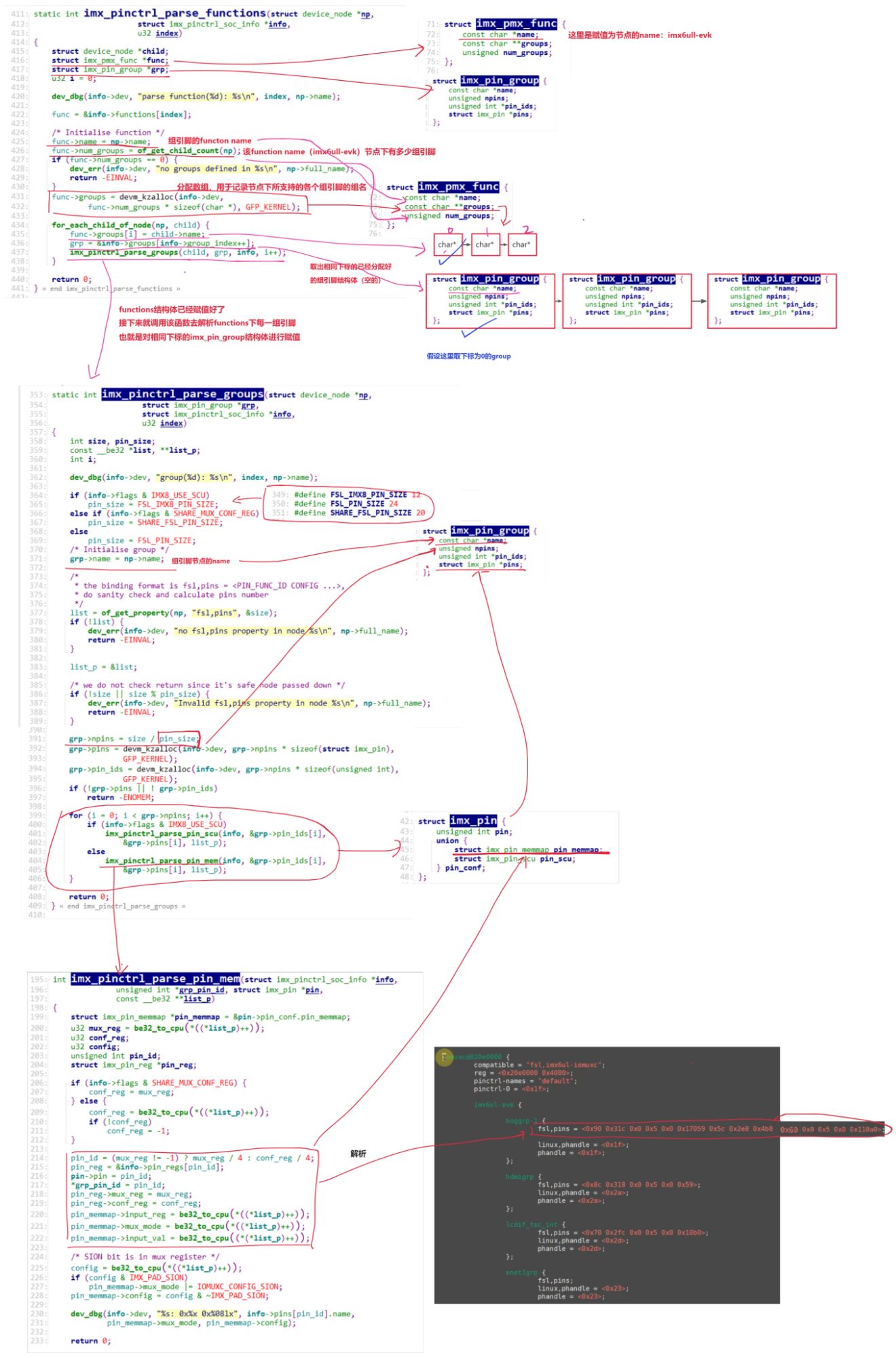
计算生物学实验5: Calling short variants with GATK4
1. 实验目的
本实验目的是利用 GATK4 工具准确高效地检测出基因组中的短变异。通过该工具对样本基因组进行分析,旨在发现单核苷酸变异(SNV)和小的插入缺失(Indel)等短变异类型。同时利用FreeBayes工具进行变异的发现,对两种工具的结果进行比较,能更好的认识发现变异的数据分析的过程。
2. 实验准备
2.1 实验平台
Linux JSvr02 3.10.0-1160.80.1.el7.x86_64 #1 SMP Tue Nov 8 15:48:59 UTC 2022 x86_64 x86_64 x86_64 GNU/Linux
2.2 数据简述
D. melanogaster WGS paired-end Illumina data:
| Data | Sample | Source |
|---|---|---|
| SRR1663608 | ZW155 | NCBI |
| SRR1663609 | ZW177 | NCBI |
| SRR1663610 | ZW184 | NCBI |
| SRR1663611 | ZW185 | NCBI |
2.3 软件配置
| 软件 | 版本 |
|---|---|
| GATK | v4.1.9.0 |
| FreeBayes | v1.3.5 |
| IGV | IGV_2.18.4 |
| samtools | 1.7 |
| bwa | 0.7.17-r1188 |
| vcftools | 0.1.15 |
3. 实验内容
分别用GATK4和FreeBayes软件进行变异发现,并对两者产生的结果进行比较,给出结论。
3.1 Calling short variants with GATK4
用GATK4对数据进行变异发现,并用IGV可视化。
(a) Fetch input files and scripts to your local scratch directory
cd /workdir
mkdir s20223282034
cd s20223282034
mkdir tmpcp /work/cb-data/Variants_workshop_2020/*.fastq.gz .
cp -r /work/cb-data/Variants_workshop_2020/genome .
cp -r /work/cb-data/Variants_workshop_2020/scripts .
tmp is a temporary directory where Java will be instructed to store its scratch files.
(b)prepare reference genome
nohup ./scripts/prepare_genome.sh >& prepare_genome.log &
- nohup 英文全称 no hang up(不挂起),用于在系统后台不挂断地运行命令,退出终端不会影响程序的运行。
Bash scripts:
#!/bin/bash# index reference genome for bwa, create fasta indexes (fai and dict)TMP=/workdir/$USER/tmp
GATKDIR=/programs/gatk-4.1.9.0
export PATH=$GATKDIR:$PATHcd genome# Genome summary files needed and by GATK tools
gatk CreateSequenceDictionary -R genome.fa -O genome.dict
samtools faidx genome.fa# index for BWA alignment
bwa index genome.fa# index image file needed by some Spark-based tools (if used)
gatk --java-options "-Djava.io.tmpdir=$TMP -Xmx4g -Xms4g" BwaMemIndexImageCreator \-I genome.fa \-O genome.fa.img
Approximate run time: 10 minutes.
result:

The files genome.fa.fai and genome.fa.dict are simple text files summarizing chromosome sizes and starting byte positions in the original FASTA file.
The other files constitute the BWA index.
©Align reads to reference
nohup ./scripts/bwa_aln.sh SRR1663608 ZW155 >& bwa_aln_SRR1663609.log &
Bash scripts:
#!/bin/bashREFFASTA=./genome/genome.fa# Un-comment one of the accession/sample pair below to run alignment for this sampleACC=$1
SAM=$2#ACC=SRR1663608
#SAM=ZW155#ACC=SRR1663609
#SAM=ZW177#ACC=SRR1663610
#SAM=ZW184#ACC=SRR1663611
#SAM=ZW185# bwa will run on 4 CPUs (-t 4)echo Alignment started
datebwa mem -M -t 4 \
-R "@RG\tID:${ACC}\tSM:${SAM}\tLB:${SAM}\tPL:ILLUMINA" $REFFASTA \
${ACC}_thinned_1.fastq.gz ${ACC}_thinned_2.fastq.gz \
| samtools view -Sb - > $ACC.bamecho Alignment finished
date
该脚本需要两个参数,即accession和sample。Approximate run time: 10 minutes .
-M: mark shorter split hits as secondary;-t:number of threads;-R: read group header line such as ‘@RG\tID:foo\tSM:bar’
Result:

(d)Sort and mark duplicates
nohup ./scripts/sort_dedup_index.sh SRR1663608 >& \ sort_dedup_index_SRR1663608.log &
Bash scripts:
# This is safer than putting them in default /tmp, which is usually smallecho Dedup/sorting started
dategatk --java-options "-Xmx16g -Xms16g" MarkDuplicatesSpark \-I ${ACC}.bam \-O ${ACC}.sorted.dedup.bam \-M ${ACC}.sorted.dedup.txt \--tmp-dir $TMP \--conf 'spark.executor.cores=4'# Separate indexing not needed if CREATE_INDEX true in MarkDuplicates#echo Indexing started
#date
此处尝试设置-Xmx4g -Xms4g,运行报错,JVM内存被耗尽。

增大内存限制,问题得以解决。Approximate run time: 10 minutes .
Check the alignment stats summary of the obtained file :
samtools flagstat SRR1663608.sorted.dedup.bam
Can you tell how many reads have been mapped? How many have been marked as duplicate?

观察结果,共比对上9606227个reads,其中740970个reads被标记为duplicates。
(e)Visualize the alignments using IGV
Use FileZilla to transfer genome.fa 、 SRR1663608.sorted.dedup.bam、SRR1663608.sorted.dedup.bam.bai to your laptop.Visualize the alignments using IGV.

选择一个片段,可以看到read是的mapping情况。
(f)Run GATK HaplotypeCaller on individual samples
nohup ./scripts/hc.sh SRR1663608 >& hc_SRR1663608.log &
Bash scripts:
#!/bin/bashREFFASTA=./genome/genome.fa
GATKDIR=/programs/gatk-4.1.9.0
#TMP=/workdir/$USER/tmp
#上述TMP位置不存在,故需修改
TMP=./tmp
export PATH=$GATKDIR:$PATHexport JAVA_HOME=/usr/local/jdk1.8.0_121
export PATH=$JAVA_HOME/bin:$PATH# We will run the genotyping on one chromosome only.
# Other chromosomes clould be handlen in separate runs,
# possibly in parallel..REGION=chr2RACC=$1# multi-threading does not work well here - they recommend using Queue to
# parallelize over regions, or just manually run a few in parallel...echo Genotyping for $ACC started
dategatk --java-options "-Xmx4g" HaplotypeCaller \--tmp-dir $TMP \-R $REFFASTA \-I $ACC.sorted.dedup.bam \-L $REGION \-ERC GVCF \--native-pair-hmm-threads 4 \--minimum-mapping-quality 30 \-O $ACC.g.vcfecho Run ended
date
To save time, we will concentrate on one chromosome, chr2R (seethe -L option in hc.sh script).
The expected result will be the file SRR1663608.g.vcf . containing the intermediate genotyping data for thissample in GVCF format.
The estimated run time of this step is 1 hour.
Result:

To save time ,fetch all the ready-made *.g.vcf files.
cp /work/cb-data/Variants_workshop_2020/premade_gvcf/*.g.vcf* .
(g) Joint variant calling with GenotypeGVCFs
nohup ./scripts/combineGVCFs.sh >& combineGVCFs.log &
bash scripts:
#!/bin/bashREFFASTA=./genome/genome.fa
GATKDIR=/programs/gatk-4.1.9.0
#TMP=/workdir/$USER/tmp
TMP=./tmpexport PATH=$GATKDIR:$PATHexport JAVA_HOME=/usr/local/jdk1.8.0_121
export PATH=$JAVA_HOME/bin:$PATH# the gVCFG files obyained before need to be combined to be used with GenotypeGVCFs toolREGION=chr2Recho Combining GVCFs started
dategatk --java-options "-Xmx4g -Xms4g" CombineGVCFs \--tmp-dir $TMP \-R $REFFASTA \-L $REGION \--variant SRR1663608.g.vcf \--variant SRR1663609.g.vcf \--variant SRR1663610.g.vcf \--variant SRR1663611.g.vcf \-O all.g.vcfecho Run ended
date
Estimated run time:5~6minutes.
Result:

To save time ,copy the pre-made *.bam and the corresponding *.bai files to your directory.
(h) Filter variants
grep "#" all.vcf > all.qual60.vcf
grep -v "#" all.vcf | awk '{if($6>60) print}' >> all.qual60.vcf
The first command extracts the VCF header lines (containing “#”) into a new VCF file, while thesecond command processes the non-header lines, appending them to the new file only if thesixth column (that’s where QUAL is) is above 60.
>>表示追加,即不清空原文件的内容,在源文件内容后面追加新的内容。
Result:

GATK offers a tool called VariantFiltration, which allows more complex filtering patterns.
nohup ./scripts/filter_vcf.sh all >& filter_vcf.log &
bash scripts:
#!/bin/bashREFFASTA=./genome/genome.fa
GATKDIR=/programs/gatk-4.1.9.0
TMP=./tmpexport PATH=$GATKDIR:$PATHexport JAVA_HOME=/usr/local/jdk1.8.0_121
export PATH=$JAVA_HOME/bin:$PATHVCF=$1 # this should be the name *without* .vcf extension!echo Run started
date# Separate snps
gatk --java-options "-Xmx4g -Xms4g" SelectVariants \--tmp-dir $TMP \-R $REFFASTA \-V $VCF.vcf \--select-type-to-include SNP \-O $VCF.snps.vcf# Filter SNPs
gatk --java-options "-Xmx4g -Xms4g" VariantFiltration \--tmp-dir $TMP \-R $REFFASTA \-V $VCF.snps.vcf \--filter-expression "QD < 2.0 || FS > 60.0 || MQ < 40.0 || ReadPosRankSum < -8.0" \--filter-name "my_snp_filter" \-O $VCF.snps.filtered.vcf# Separate indels
gatk --java-options "-Xmx4g -Xms4g" SelectVariants \--tmp-dir $TMP \-R $REFFASTA \-V $VCF.vcf \--select-type-to-include INDEL \-O $VCF.indels.vcf# Filter indels
gatk --java-options "-Xmx4g -Xms4g" VariantFiltration \--tmp-dir $TMP \-R $REFFASTA \-V $VCF.indels.vcf \--filter-expression "QD < 2.0 || FS > 200.0" \--filter-name "my_indel_filter" \-O $VCF.indels.filtered.vcf# Merge filtered files
gatk --java-options "-Djava.io.tmpdir=$TMP -Xmx4g -Xms4g" MergeVcfs \-R $REFFASTA \-I $VCF.snps.filtered.vcf \-I $VCF.indels.filtered.vcf \-O $VCF.filtered.vcf \echo Run ended
date
Result:

Note that no variant has been removed from the file. The ones that failed filtering are just marked as such. Estimated run time: 3 minutes.
3.2 Joint variant calling using FreeBayes
用FreeBayes对数据进行变异发现
conda activate FreeBayes
nohup ./scripts/fb.sh >& fb.log &
bash scripts:
#!/bin/bash#FB=/programs/freebayes/bin/freebayes
FB=freebayesREFFASTA=./genome/genome.faecho Started at
date$FB -f $REFFASTA \
SRR1663608.sorted.dedup.bam \
SRR1663609.sorted.dedup.bam \
SRR1663610.sorted.dedup.bam \
SRR1663611.sorted.dedup.bam \
--min-mapping-quality 30 \
-r chr2R > fb.vcfecho Completed at
date
error:

经查询错误信息,认为是samtools出现问题,尝试解决,建立软链接:
ln -s /work/miniconda3/lib/libcrypto.so.1.1 /work/miniconda3/lib/libcrypto.so.1.0.0

权限不足,请求被拒绝。
3.3 Basic stats and comparison of variant sets
对发现的变异进行统计,并对两种不同软件发现的变异进行比较。
(a) Using Linux commands
grep -v "#" all.vcf | wc -l
#result:423493
grep -v "#" all.vcf | awk '{if($1=="chr2R" && $2 >=10000 && $2 <=20000) print}' >extracted_records
awk '{if($7=="PASS") print}' all.filtered.vcf | wc -l
#result:417825
(b) Using GATK4’s Concordance and GenotypeConcordance tools
./scripts/var_compar.sh fb all ZW155 >& var_compar.log &
bash scripts:
#!/bin/bashREFFASTA=./genome/genome.fa
GATKDIR=/programs/gatk-4.1.9.0
TMP=/workdir/$USER/tmpexport PATH=$GATKDIR:$PATHexport JAVA_HOME=/usr/local/jdk1.8.0_121
export PATH=$JAVA_HOME/bin:$PATH# Supply only core file names (i.e., without .vcf suffix) of the VCF files
VCF1=$1
VCF2=$2
SAMPLE=$3# variants in VCF2 will be called "truth", and those from VCF1
# will be "evaluated" against them.# Check site concordance between two variant sets
gatk --java-options "-Xmx2g -Djava.io.tmpdir=$TMP" Concordance \-R $REFFASTA \--evaluation $VCF1.vcf \--truth $VCF2.vcf \--summary $VCF1.$VCF2.comp.site_summary# Check genotype concordance for sample SAMPLE
gatk --java-options "-Xmx2g -Djava.io.tmpdir=$TMP" GenotypeConcordance \--CALL_VCF $VCF1.vcf \--TRUTH_VCF $VCF2.vcf \--CALL_SAMPLE $SAMPLE \--TRUTH_SAMPLE $SAMPLE \--O $VCF1.$VCF2.comp
由于FreeBayes发现变异的步骤失败,故无结果。
© Using vcftools
-
Obtain basic VCF statistics (number of samples and variant sites):
vcftools --vcf all.vcf --freq -c > all.freqsresult:423493 (same to using linux commands)

-
Extract subset of variants (chromosome chr2R, between positions 1M and 2M) and write them toa new VCF file
vcftools --vcf all.vcf --chr chr2R --from-bp 1000000 --to-bp 2000000 --recode -- recode-INFO-all -c > subset.vcfResult:

-
Get allele(等位基因) frequencies for all variants and write them to a file
vcftools --vcf all.vcf --freq -c > all.freqsResult:

-
Compare two VCF files
vcftools --vcf fb.vcf --diff all.vcf --out fb.all.compare由于没有
fb.vcf,无结果。
4. 实验总结
4.1 实验结论
GATK4 能够有效地识别样本基因组中的单核苷酸变异和小的插入缺失等短变异类型。该工具具有较高的准确性和可靠性,为后续的疾病研究和遗传分析提供了坚实的数据基础。同时,实验过程也表明,正确的参数设置和数据预处理对于获得高质量的结果至关重要。
4.2 实验收获
学会了GATK4工具和FreeBayes工具的使用,并复习了许多Linux的命令操作,对变异发现的过程有了较好的理解。
4.3 其它心得
实践过程中,可以多查阅分析软件的官方文档,这样既能提高英语阅读能力,也能更正确的认识这些软件。
d write them to a file
vcftools --vcf all.vcf --freq -c > all.freqs
Result:
[外链图片转存中…(img-APRbfE5A-1729756791664)]
-
Compare two VCF files
vcftools --vcf fb.vcf --diff all.vcf --out fb.all.compare由于没有
fb.vcf,无结果。
4. 实验总结
4.1 实验结论
GATK4 能够有效地识别样本基因组中的单核苷酸变异和小的插入缺失等短变异类型。该工具具有较高的准确性和可靠性,为后续的疾病研究和遗传分析提供了坚实的数据基础。同时,实验过程也表明,正确的参数设置和数据预处理对于获得高质量的结果至关重要。
4.2 实验收获
学会了GATK4工具和FreeBayes工具的使用,并复习了许多Linux的命令操作,对变异发现的过程有了较好的理解。
4.3 其它心得
实践过程中,可以多查阅分析软件的官方文档,这样既能提高英语阅读能力,也能更正确的认识这些软件。