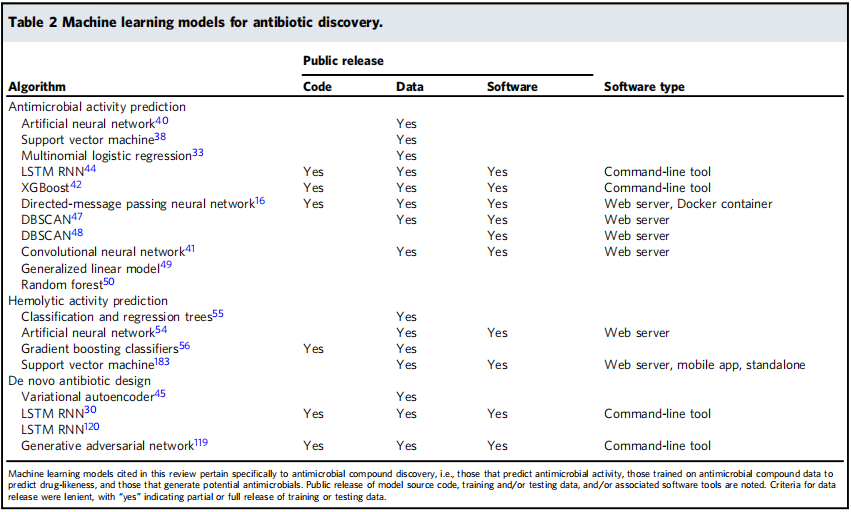
目录
- 基于AI的靶点识别概述
- 肿瘤建模
- 肿瘤转录组
- 转录组异质性
- 单细胞转录组数据分析
- 单细胞表观肿瘤模型
- 肿瘤中的表观遗传模型
- 基于甲基化测序技术的肿瘤表观遗传模型
- 基于染色质可及性的肿瘤表观遗传模型
- 多模态肿瘤建模
- 靶点识别
- 基于单细胞RNA的靶点发现
- 基于表观的靶点发现
- 基于多组学的靶点发现
- 总结
基于AI的靶点识别概述
肿瘤药物研发是AIDD的应用场景,前提是靶点识别,早期靶点识别以肿瘤细胞的高变基因的致癌蛋白为主,筛选药物靶向这些蛋白。经过长时间的临床测试,发现这种方案覆盖的患者群体很有限。飞速发展的组学测量技术为发现新靶点带来希望。
- 组学通常指生物学中对各类研究对象(一般为生物分子)的集合所进行的系统性研究,比如基因组,蛋白质组,转录组等。
- 传统的批量组学技术(bulk)是以个体为研究对象,将待测生物样本中所有细胞混合在一起进行分子测定,只能反映两个肿瘤样本个体间的差异。
- 现在的单细胞组学技术是对肿瘤样本中每个细胞进行分子的含量测量,全面刻画肿瘤细胞间以及肿瘤免疫微环境的异质性,为发现新的肿瘤靶点提供了更详细的方案。
随着测量技术的普及,肿瘤细胞的批量组学和单细胞组学数据都在快速积累。但是这些组学数据都有一个共同挑战:维度高,噪声大,数据类型多样等特点。
肿瘤建模
肿瘤转录组
转录组异质性
癌症的普遍特点是转录失调。在细胞内部,调节网络由一组连接的途径组成,其中途径是细胞中发生的化学反应链,通路是基因的集合,这些基因相互作用可以实现特定的细胞功能。为了使细胞正常运作,通路基因的表达水平需要得到控制。
- 正常细胞和癌细胞存在很多差异表达的基因,癌细胞中的异常表达能通过抑制或刺激途径使途径失调,这可能会影响细胞的增殖能力,这种转录组上的差异即为癌症中的转录组异质性。
转录组指的是细胞内所有转录产物的集合,包括信使RNA,核糖体RNA,转运RNA,非编码RNA。
转录组异质性在癌细胞中非常明显,这源于DNA拷贝的异常,细胞所处环境的刺激,基因相互作用混乱等(对于基因组相同的细胞,由于上述原因导致表现出不同的转录状态)。从转录层面看,癌症是一种细胞调节网络混乱的疾病,因此转录组的研究可以更好探索癌细胞。单细胞RNA测序可以绘制细胞的转录图谱,清晰展现细胞的转录特点。
近年来,单细胞RNA测序(scRNA-seq)得到广泛应用,使得我们在单细胞水平研究转录组。2009年首次出现scRNA-seq方法,随后出现测量技术的改进,比如Drop-seq,Seq-Well,DroNC-seq,SPLiT-seq等。其中,基于液滴的技术(Drop-seq,InDrop,Chromium)可以提供更大的细胞通量,而且与全转录scRNA-seq相比,测量成本更低,因此更被推荐。另外,商业化技术以10xGenomics为主。
单细胞转录组数据分析
质量控制:scRNA-seq数据技术噪声水平高,破损,死亡或与多个细胞混合的细胞中会生成低质量数据,这些低质量数据阻碍下游分析,因此,需要质量控制。
批次校正:测序过程中的操作差异,平台差异,测序方法差异等会引入系统误差,导致一个批次的基因表达谱与另一个批次的基因表达谱存在系统误差,这种批次差异可能会掩盖真实的生物学差异,导致分析错误,因此需要批次校正。现在广泛使用的批次校正方法为Harmony,LIGER,Seurat v3。
插补去噪:scRNA-seq数据包含许多由于原始RNA扩增失败导致的dropouts,因此需要插补算法进行去噪:SAVER,MAGIC,ScImpute,DrImpute,AutoImpute。其中,SAVER利用基于UMI的scRNA-seq恢复所有基因的表达,MAGIC构建基于马尔可夫亲和度的基因表达图来计算基因表达,ScImpute利用类似细胞中dropout影响较小的相同基因信息,恢复dropout值。
降维:由于数据是高维的,我们需要降维和特征提取算法进一步分析数据。主流降维算法分为:基于矩阵分解,基于图,基于神经网络。分别对应常用的三类方法:PCA,t-SNE,UMAP。PCA是最常用的线性降维方法。t-SNE根据在邻域图上随机游走的概率分布在数据中找到结构关系。UMAP是基于k-近邻使用随机梯度下降优化的结果。
单细胞表观肿瘤模型
肿瘤中的表观遗传模型
染色质结构定义了DNA形式的遗传信息在细胞内的状态,基因组的结构影响着基因被激活或沉默的能力。表观遗传学被定义为:基因及其产物之间的因果相互作用,从而导致表型的形成,涉及到理解染色质结构及其对基因功能的影响。现代表观遗传学研究独立于原始DNA序列变化而发生的基因表达的可遗传变化。
这些可遗传的变化大部分在分化过程中建立,并在细胞分裂的多个周期中维持,使细胞在包含相同遗传信息的同时具有不同的身份。这是由表观遗传修饰决定的,包括DNA中胞嘧啶碱基的甲基化,组蛋白的翻译后修饰,核小体沿DNA的定位。这些修饰的补充,统称为表观基因组。如果不能正确维护可遗传的表观遗传标记,可能导致各种信号通路的不适当激活或抑制,导致癌症等疾病。
与基因突变不同,表观遗传畸变具有潜在可逆性,因此可以通过找到表观遗传层面肿瘤的靶点信息,治疗患者,使癌细胞恢复正常。第二代测序技术的进步,推动了表观遗传学的发展。例如:
- ChIP-seq技术可以绘制组蛋白修饰和转录因子结合的全基因组图谱。
- 利用ATAC-seq可以测定染色质可及性。
- Hi-C可以确定高阶染色质结构。
- DNA甲基化可以通过RRBS,WGBS或基于阵列的技术来确定。
生物表观遗传辅助诊前景广阔,但当前生物表观遗传辅助诊断面临许多挑战,比如数据分布广泛,模态多。我们希望借助机器学习技术集成大型数据,推动诊断的发展,帮助医生进一步解析数据。
基于甲基化测序技术的肿瘤表观遗传模型
基因甲基化使表观遗传的主要形式,同癌症的发生与发展有重要联系。DNA甲基化代表基因组的直接修饰,并调控基因表达。相比正常细胞,癌细胞的基因甲基化水平显著下降,这是癌细胞基因调控失衡的重要原因。
对于正常组织,细胞通过基因甲基化实现表达调控,当相关基因调控区的CpG岛大量发生甲基化时,会阻止该基因表达(即沉默)。DNA甲基化导致基因沉默的机制大致分三种:
- DNA甲基化干扰转录因子对DNA元件的识别与结合;
- 序列特异性的甲基化DNA结合蛋白与启动子区甲基化CpG岛结合,形成转录抑制复合物,阻止转录因子与启动子区的结合;
- DNA甲基化改变染色质结构,使染色质更加紧密,影响转录因子与DNA结合。
mSEPT9基因甲基化是结直肠癌的生物标记物,mSEPT9基因编码的SEPT9蛋白在细胞代谢中发挥作用。SEPT9蛋白可阻止细胞过快分裂,具有抑癌作用。当SEPT9启动子区域甲基化时,SEPT9蛋白停止表达,导致上皮细胞癌变,发展为结直肠癌。
基因甲基化靶点的发现依赖算法,关键在于从噪声数据中识别出同癌症发生相关度高的基因甲基化信号,存在下面挑战:
- 基因甲基化数据混合了各癌症克隆亚型以及正常细胞型的甲基化信号;
- 单一基因的表达静默或开启存在多种甲基化模式;
- 数据维度高,样本量有限;
机器学习在表观遗传领域通常是建立一个模型,准确预测给定样本的类别信息,例如从矩阵数据中区分正常与癌症样本。
针对高维数据挑战,现有工作从特征排序+特征选择和特征融合三种思路进行降维。特征排序通过假设检验检测特征与标签的关联,并对特征的重要度进行排名。例如T检验计算P值来衡量零假设,即潜在假设是患者样本和对照组样本都符合正态分布。Wilcoxon检验评估两个分布之间的差异,其作为T检验的替代。特征选择建立在特征排序基础上,并考虑筛选特征。特征融合将多特征融合为单特征,以降低特征数量。
基于染色质可及性的肿瘤表观遗传模型
单细胞染色质可及性是在单细胞水平检测基因所在染色体是否处于开放状态:处于染色质开放状态的基因可被转录并表达,处于染色质闭合状态的基因会沉默。ATAC通常与DNA甲基化相关。相比甲基化测序,ATAC提供了更直接和确定的观测。
相比scRNA-seq,scATAC-seq分析更困难:
- scATAC-seq高度稀疏,当前scATAC-seq测序技术仅能覆盖1%的基因组,此外,测量信号微弱,仅能测量极少数染色质的打开基因。
- 研究浅薄,数据库极度缺乏。
- 数据维度更高,scRNA-seq侧重于测量蛋白编码基因的RNA表达量,大约3万。scATAC-seq不局限于测量蛋白编码基因区间的染色质开合状态,测量范围包括全基因组。
多模态肿瘤建模
多组学分析,是指同时在多种不同的生物分子层面对研究对象进行分析,通过将基因组,表观组,转录组,蛋白组,代谢组等不同模态的分子数据整合,揭示更丰富的生物规律。单细胞多模态技术将肿瘤多组学分析的分辨率提高到细胞水平。
癌症多组学模型从信息整合方式上来看分为早期整合,中期整合,后期整合。见下图。最常用的是中期整合,它避免了早期整合的维度不匹配或样本不匹配问题,以及避免了后期整合的整合前分析信息不完备问题。

AI在单细胞多组学领域的应用场景分两种:
- 第一种场景是不同组学的测量对象(细胞)不匹配。通常,假设每种组学测量的细胞来自同一个分布。
- 第二种场景是不同组学的测量对象是同一批细胞。
靶点识别
基于单细胞RNA的靶点发现
以RNA失调作为肿瘤治疗的入口,寻找新的免疫治疗靶点成为肿瘤研究的热点,对不同RNA亚型以及参与RNA加工的蛋白质促成癌症的机制发现,为治疗癌症带来机遇。比如circRNA在癌症中过度表达展示了其作为疾病生物标志物的潜力。
转录组可用于肿瘤及免疫细胞的亚群分析,为肿瘤发展,免疫逃逸和耐药性研究提供全面信息。转录组层面的亚群分析可以提供亚群独特的蛋白等标志物,进而针对特定肿瘤亚群,免疫抑制性细胞亚群指定靶点药物。
从转录组出发的肿瘤细胞之间的相互作用研究也有望提供靶点。当前细胞间通信的识别方法有两种:
- 依赖于一种细胞类型中受体基因与另一种细胞类型中相应配体基因的表达水平比较。CellPhoneDB方法首先计算一种细胞类型中受体基因的平均表达和另一种细胞类型中配体基因的平均表达,然后通过基于图的方法生成零分布,以评估统计显著性。
- 计算一种细胞类型的受体基因表达与另一种细胞类型中相应配体基因表达在所有scRNA-seq数据集中的相关性识别特定的通信。
基于表观的靶点发现
表观调控异常是肿瘤细胞恶化的原因。从表观调控异常信号中搜索癌症的生物标记物具有广阔的临床应用前景,可用于开发能逆转肿瘤表观遗传异常的药物,抑制癌细胞增殖。
基于表观组学分析的人工智能方法可用于癌症亚型分类,协助指定个性化临床治疗方案。另一种用途是肿瘤-健康细胞差异分析和靶点识别。
基于多组学的靶点发现
肿瘤的发生是自身异常基因突变积累(内因)与免疫系统失衡(外因)共同导致的。因此,肿瘤治疗有两种思路:
- 针对肿瘤细胞的异常特征识别出可以直接作用于肿瘤细胞的靶点。
- 根据肿瘤免疫微环境的特点,识别作用于免疫细胞的靶点,从而提升免疫细胞对肿瘤的抑制效果。
其中,第二种方法即2018年获得诺贝尔奖的免疫疗法:利用患者自身免疫系统来预防,控制和消除癌症。免疫系统本身具备识别并消灭肿瘤细胞的功能,然而在肿瘤不断演化的过程中,肿瘤细胞学会了避开免疫系统的杀伤,从而产生免疫逃逸。
充分解析肿瘤免疫微环境,是发现免疫靶点的重要基础和前提。研究表明,肿瘤微环境会将T细胞长期暴露于抗原的持续慢性刺激下,使得T细胞丧失效应功能。T细胞失活导致细胞状态改变,在表观组,转录组和代谢组等方面均会体现异常。
目前,人们对肿瘤免疫微环境的了解有限,充分了解微环境的细胞构成,认识每种细胞类型在肿瘤发生过程中的作用,有望提升现有的免疫疗法水平。
AI在解构微环境上具有潜力。北京大学对肺癌免疫微环境中的T细胞群体进行了多模态分析,发现除肿瘤相关的CD8 T细胞呈现耗竭状态外,还有两种细胞呈现耗竭状态,并识别出对应的基因标志。
总结
AI结合单细胞数据,可以辅助靶点识别,从而进行药物筛选。此外,单细胞组学可细致刻画药物临床实验中的实验组和对照组在不同分子层面的差异,辅助判定药物的有效性,揭示药物作用机制。