文章目录
- 一、实验介绍
- 二、实验环境
- 1. 配置虚拟环境
- 2. 库版本介绍
- 三、实验内容
- 0. 导入必要的库
- 1. 读取数据集
- 2. 质量控制(可选)
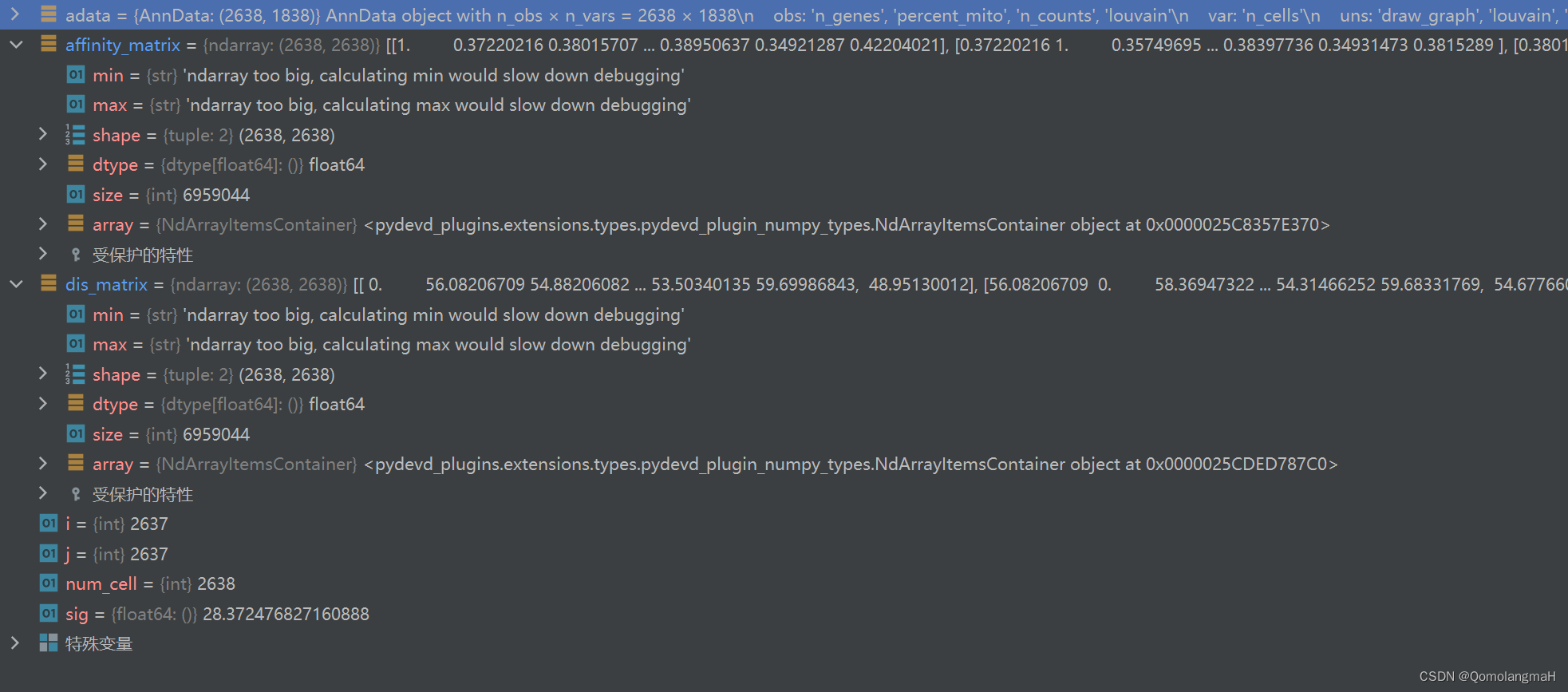
- 3. 基于距离的亲和力矩阵
- 4. 绘制基因表达的Heatmap
- 5. 基于皮尔逊相关系数的亲和力矩阵
- 6. 代码整合
一、实验介绍
计算亲和力矩阵,一般按照以下步骤进行:
- 导入数据:加载单细胞RNA测序数据集。
- 数据预处理:根据需要对数据进行预处理,例如基因过滤、归一化等。
- 计算亲和力:使用合适的算法(例如,欧几里德距离、Pearson相关系数或其他距离/相似度度量)计算样本之间的亲和力(可以使用现有的生物信息学工具包(如Scanpy)来执行此计算。
- 构建亲和力矩阵:将计算得到的亲和力值组织成一个亲和力矩阵,其中每个元素表示两个样本之间的亲和力。
二、实验环境
1. 配置虚拟环境
可使用如下指令:
conda create -n bio python=3.8
conda activate bio
pip install -r requirements.txt
其中,requirements.txt:
numpy==1.18.1
matplotlib==3.1.2
seaborn==0.9.02. 库版本介绍
| 软件包 | 本实验版本 | 目前最新版 |
|---|---|---|
| matplotlib | 3.1.2 | 3.8.0 |
| numpy | 1.81.1 | 1.26.0 |
| python | 3.8.16 | |
| scipy | 1.10.1 | 1.11.3 |
| seaborn | 0.12.2 | 0.13.0 |
三、实验内容
0. 导入必要的库
import scanpy as sc
import numpy as np
from scipy.spatial import distance_matrix
import seaborn as sns
import matplotlib.pyplot as plt
from scipy.stats import pearsonr
- Scanpy是一个用于单细胞RNA测序数据分析的Python库,提供了许多功能和工具来处理和分析单细胞数据
1. 读取数据集
adata = sc.read_h5ad('./pbmc3k.h5ad')
在生物信息学中,PBMC3K.h5ad是一种常用的单细胞RNA测序数据集,用于研究人类外周血单个核细胞(PBMC)的基因表达。
2. 质量控制(可选)
# 质控
# 过滤掉低质量的细胞和基因
sc.pp.filter_cells(adata, min_genes=200) # 过滤掉表达基因数目小于200的细胞
sc.pp.filter_genes(adata, min_cells=30) # 过滤掉被少于3个细胞表达的基因3. 基于距离的亲和力矩阵
import scanpy as sc
import numpy as np
from scipy.spatial import distance_matrix# 计算亲和力矩阵
adata = sc.read_h5ad('./pbmc3k.h5ad')
dis_matrix = distance_matrix(adata.X, adata.X) # calculate distance matrix
num_cell = dis_matrix.shape[0]
sig = np.var(dis_matrix) # sigma
affinity_matrix = np.zeros((num_cell, num_cell))
for i in range(num_cell):for j in range(num_cell):affinity_matrix[i,j] = np.exp(-dis_matrix[i,j] /(2 * sig))
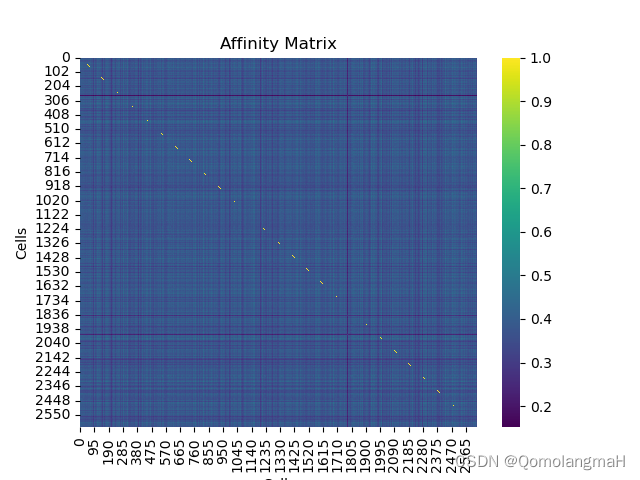
4. 绘制基因表达的Heatmap
sns.heatmap(affinity_matrix, cmap='viridis')
plt.title('Affinity Matrix')
plt.xlabel('Cells')
plt.ylabel('Cells')
plt.show()
ChatGPT:
热图(Heatmap)是一种数据可视化技术,用于显示数据中的密度和模式。它通过将数据点映射到颜色编码的图像上来展示数据的分布情况。热图通常用于显示二维数据,其中每个数据点的位置对应于平面上的坐标,并使用颜色来表示数据点的密度或值。
在一个热图中,颜色编码表示了数据点的频率或强度。通常,较高的频率或强度用较亮或较暖的颜色(如红色)表示,而较低的频率或强度用较暗或较冷的颜色(如蓝色)表示。这种颜色映射使得我们能够直观地观察和分析数据的分布特征,从而揭示出数据集中的模式、热点和趋势。
热图在多个领域和应用中都得到了广泛使用。在数据分析和可视化中,热图常用于显示热点地区、人口密度、温度分布、点击热度、基因表达模式等。在商业领域,热图可以帮助用户更好地理解和解释数据,从而支持决策制定和问题解决。此外,热图还在医学、生物学、交通规划、市场营销等领域中发挥着重要作用。
5. 基于皮尔逊相关系数的亲和力矩阵
【生物信息学】使用皮尔逊相关系数进行相关性分析
from scipy.stats import pearsonr
# 计算每对细胞之间的皮尔逊相关系数pearson_matrix = np.zeros((num_cell, num_cell))
for i in range(num_cell):for j in range(num_cell):pearson_matrix[i, j] = pearsonr(adata.X[i], adata.X[j])[0]# 将合并的亲和力矩阵保存
adata.obsp['distances'] = combined_affinitysns.heatmap(combined_affinity, cmap='viridis')
plt.title('Combined Matrix')
plt.xlabel('Cells')
plt.ylabel('Cells')
plt.show()
6. 代码整合
import scanpy as sc
import numpy as np
from scipy.spatial import distance_matriximport seaborn as sns
import matplotlib.pyplot as plt
from scipy.stats import pearsonr# 计算亲和力矩阵
adata = sc.read_h5ad('./pbmc3k.h5ad')
dis_matrix = distance_matrix(adata.X, adata.X) # calculate distance matrix
num_cell = dis_matrix.shape[0]
sig = np.var(dis_matrix) # sigma
affinity_matrix = np.zeros((num_cell, num_cell))
for i in range(num_cell):for j in range(num_cell):affinity_matrix[i,j] = np.exp(-dis_matrix[i,j] /(2 * sig))# %%# 创建热图
sns.heatmap(affinity_matrix, cmap='viridis')
plt.title('Affinity Matrix')
plt.xlabel('Cells')
plt.ylabel('Cells')
plt.show()# %%
from scipy.stats import pearsonr
# 计算每对细胞之间的皮尔逊相关系数pearson_matrix = np.zeros((num_cell, num_cell))
for i in range(num_cell):for j in range(num_cell):pearson_matrix[i, j] = pearsonr(adata.X[i], adata.X[j])[0]# 将基于距离的亲和力矩阵和皮尔逊相关系数亲和力矩阵相加
combined_affinity = affinity_matrix + pearson_matrix# 将合并的亲和力矩阵保存
adata.obsp['distances'] = combined_affinitysns.heatmap(combined_affinity, cmap='viridis')
plt.title('Affinity Matrix')
plt.xlabel('Cells')
plt.ylabel('Cells')
plt.show()