作者:Squirrelity
2022-07-18 补充说明
最近R更新了,很多包都用不了,如果遇到报错或者是运行不了有可能是因为版本问题。
一、加载对应的R包
这里用到十三个包(距离上次更新之后又新增了不少方法/包):
library("TCGAbiolinks")
library("plyr")
library("limma")
library("biomaRt")
library("SummarizedExperiment")
library("stringr")
library("ggplotify")
library("patchwork")
library("cowplot")
library('DESeq2')
library('edgeR')
library("dplyr")
library("rtracklayer")
光下载都费了不少功夫www,下面把install代码放出来(。・∀・)ノ゙直接install.packages()日常失败我就不说了
大部分包都可以在bioconductor中找到,有遗漏的可以去官网找下载代码
https://bioconductor.org/
#plyr下载 唯一可以直接install的包啊哈哈
install.packages("plyr")
#if(!require(stringr))
#install.packages()下载法
if(!require(stringr))install.packages('stringr')
if(!require(ggplotify))install.packages("ggplotify")
if(!require(patchwork))install.packages("patchwork")
if(!require(cowplot))install.packages("cowplot")#更新BiocManager3.15,R版本为4.2.0
if (!require("BiocManager", quietly = TRUE))install.packages("BiocManager")
BiocManager::install(version = "3.15")#tccgbiolinks包稳定版本安装
if (!requireNamespace("BiocManager", quietly=TRUE))install.packages("BiocManager")
BiocManager::install("TCGAbiolinks")#limma包下载
if (!require("BiocManager", quietly = TRUE))install.packages("BiocManager")
BiocManager::install("limma")#biomaRt包下载
if (!require("BiocManager", quietly = TRUE))install.packages("biomaRt")
BiocManager::install("biomaRt")#最麻烦的SummarizedExperiment包:force = TRUE是根据warning后来加的if (!require("BiocManager", quietly = TRUE))install.packages("BiocManager")
BiocManager::install("SummarizedExperiment",force = TRUE)
#edgeR包下载
if (!require("BiocManager", quietly = TRUE))install.packages("BiocManager")
BiocManager::install("edgeR")
#DESeq2包下载
if (!requireNamespace("BiocManager", quietly=TRUE))install.packages("BiocManager")
BiocManager::install("DESeq2")
在使用之前需要加载BIocManager,代码参考这个:https://bioconductor.org/install/
if (!require("BiocManager", quietly = TRUE))install.packages("BiocManager")
BiocManager::install(version = "3.15")
若提示warning:A version of this package for your version of R might be available elsewhere,see the ideas at
https://cran.r-project.org/doc/manuals/r-patched/R-admin.html#Installing-packages
可以的解决方法有:
1.乖乖在官网下载cran(最不推荐)
2.在Rstudio上方的tools-global options处找到packages,修改默认的global cran,选择apply-ok即可
3.找到包的官网,用官网提供的代码下载(首推♥♥♥)
二、下载数据
在下载之前设置工作路径:
dir.create()创建目录,getwd()获取工作路径,setwd()设置工作路径,由于TCGA下载下来的数据包都挺大的,建议还是选一个比较富裕的盘来作为工作路径。
dir.create("D:\\BioInfoCloud\\TCGABiolinks\\case_study")
setwd("D:\\BioInfoCloud\\TCGABiolinks\\case_study")
这里用到的是R包TCGAbiolinks:
可以参照R包TCGAbiolinks官网使用http://www.bioconductor.org/packages/release/bioc/vignettes/TCGAbiolinks/inst/doc/casestudy.html#Case_study_n_1:_Pan_Cancer_downstream_analysis_BRCA
示例:
query <- GDCquery(project = "TCGA-BRCA",data.category = "Transcriptome Profiling",data.type = "Gene Expression Quantification", workflow.type = "STAR - Counts")
project选出的是肿瘤项目,而里面用到的都是缩写,详见https://blog.csdn.net/Squirrelity/article/details/124259330?spm=1001.2014.3001.5501
建议去TCGA官网repository一边对照着选所需要的样本
https://portal.gdc.cancer.gov/repository?facetTab=cases
#下载到本地
GDCdownload(query = query, method = "api")
#查看下载的数据
View(query)
BRCA.Rnaseq.SE <- GDCprepare( query = query, save = TRUE, save.filename = "brca.rda")
BRCAMatrix <- assay(BRCA.Rnaseq.SE,"unstranded")
#记住这个文件BRCA.RNAseq_CorOutliers
BRCA.RNAseq_CorOutliers <- TCGAanalyze_Preprocessing(BRCA.Rnaseq.SE)三、ID转换
下载下来的BRCA.RNAseq_CorOutliers为ENTREZID,而我们肯定是需要图片显示基因名而不是ENTREZID,因此进行数据转换,这里用到包括但不限于的dplyr包和rtracklayer包
library("dplyr")
library("rtracklayer")
ID转换分为四步:
1.导入数据:BRCA.RNAseq_CorOutliers和人类基因组注释文件;
data=read.table(BRCA.RNAseq_CorOutliers,header=T,sep='\t')
#把行名改为gene_id,与gtf保持一致
colnames(data)[1] <- "gene_id"
对照人类基因组注释文件,对BRCA.RNAseq_CorOutliers进行ID转换
其中,人类基因组注释文件参考http://www.360doc.com/content/21/1028/10/77506210_1001626502.shtml
#处理人类基因组注释文件的数据
gtf <- rtracklayer::import('Homo_sapiens.GRCh38.99.chr.gtf.gz')
gtf <- as.data.frame(gtf)
save(gtf,file="人类基因组注释文件.Rda")
gtf <-load(file="人类基因组注释文件.Rda")
#根据条件筛选基因(大筛选)
a = dplyr::filter(gtf,type=="gene")
dim(a)
#只要gene_name,gene_id,gene_biotype这三行
b = dplyr::select(a,c(gene_name,gene_id,gene_biotype))
2.数据预处理
#ENTREZID带有,这里去除小数点及后边的数字(我用excel处理的,ctrl+F无字符替换.*)
data1 <- separate(data,gene_id,into = c("gene_id"),sep="\\.")
3.数据处理
#根据gene id 合并文件
c = dplyr::inner_join(b,data,by="gene_id")
#去掉2,3列,基因名再去重
d=select(c,-gene_id,-gene_biotype)
data1=distinct(d,gene_name,.keep_all = T)
#把行名由数字换成基因
rownames(data1)<- data1[,1]
data1<-data1[,-1]
4.数据后处理
#如下载的数据取了log2(count-1)这里再返回count
data2 <- 2^data1 -1
write.csv(data2,file="data2.csv")
data2 <- read.csv("data2.csv")
#重新用read打开整行的-会变成.因此需要恢复原来的行名
colnames(data2) <- colnames(BRCA.RNAseq_CorOutliers)
四、差异表达分析
1.参考网址
代码示例参考这个包的文档http://bioconductor.org/packages/release/bioc/html/TCGAbiolinks.html
TCGA可视化教程
https://www.jianshu.com/p/d3e481f0187a
https://cloud.tencent.com/developer/article/1778874
http://bioconductor.org/packages/release/bioc/vignettes/TCGAbiolinks/inst/doc/casestudy.html
2.数据预处理
#处理后可得热图判断样本相似性
dataPrep <- TCGAanalyze_Preprocessing(object = BRCA.RNAseq_CorOutliers, cor.cut = 0.6
)
*上面代码生成的图如下所示。组内的样本相似性都很高,符合预期。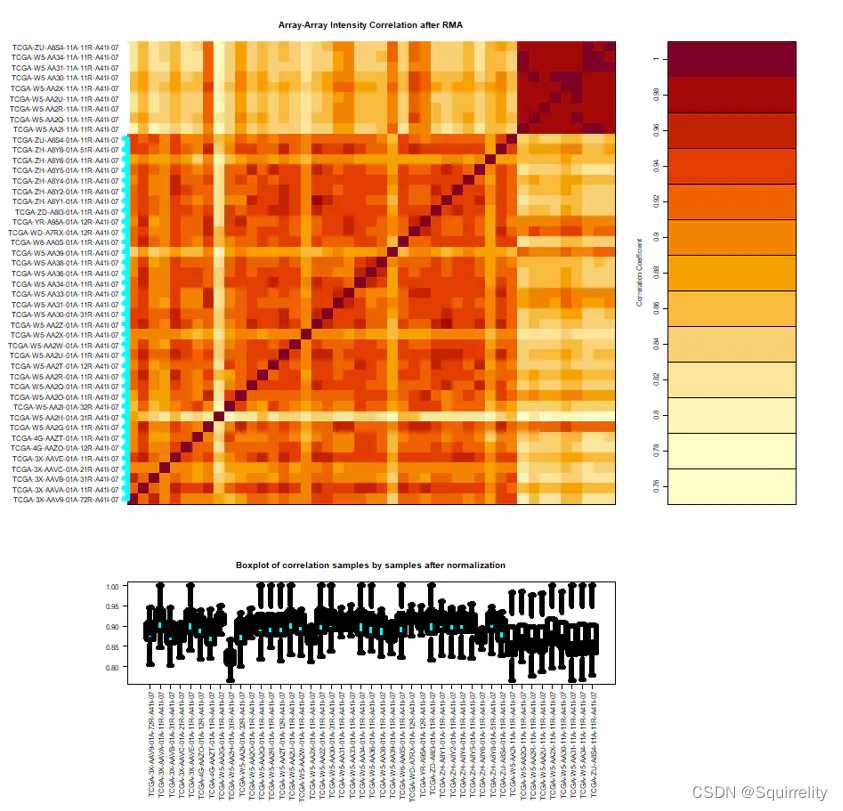
3.对数据进行标准化处理+质控+差异化分析
**TCGAanalyze_LevelTab()**将差异表达基因在正常和肿瘤组织中的表达量数据添加到差异表达分析结果中的主要用法:
TCGAanalyze_LevelTab(FC_FDR_table_mRNA, typeCond1, typeCond2, TableCond1,
TableCond2, typeOrder = TRUE)

#数据标准化
dataNorm <- TCGAanalyze_Normalization(tabDF = data2,geneInfo = geneInfoHT,method = "gcContent"
)
#数据质控
dataFilt <- TCGAanalyze_Filtering(tabDF = dataNorm,method = "quantile", qnt.cut = 0.25
) #分组:NT正常组织组 TP癌症组织组*# selection of normal samples "NT"
samplesNT <- TCGAquery_SampleTypes(barcode = colnames(dataFilt),typesample = c("NT")
))
# selection of tumor samples "TP"
samplesTP <- TCGAquery_SampleTypes(barcode = colnames(dataFilt), typesample = c("TP")
)# 差异表达分析
dataDEGs <- TCGAanalyze_DEA(mat1 = dataFilt[,samples.solid.tissue.normal],mat2 = dataFilt[,samples.primary.tumour],Cond1type = "Normal",Cond2type = "Tumor",fdr.cut = 0.01 ,logFC.cut = 2,method = "glmLRT",pipeline = "edgeR"
) #在正常和肿瘤样本中差异基因的表达值
dataDEGsFiltLevel <- TCGAanalyze_LevelTab(FC_FDR_table_mRNA = dataDEGs,typeCond1 = "Tumor",typeCond2 = "Normal",TableCond1 = dataFilt[,samplesTP1],TableCond2 = dataFilt[,samplesNT1]
)
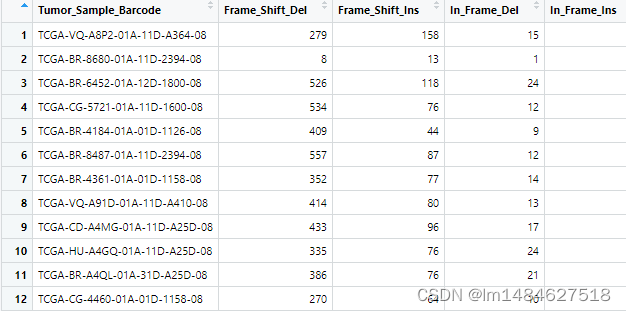
得到的dataDEGsFiltLevel文件按logFC绝对值排序可得最显著的top差异表达基因(excel表处理)
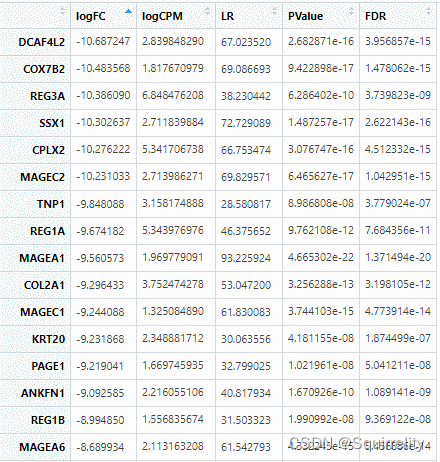
五、可视化
ps:得到的图片有的可以直接看,有的保存在工作路径上了。
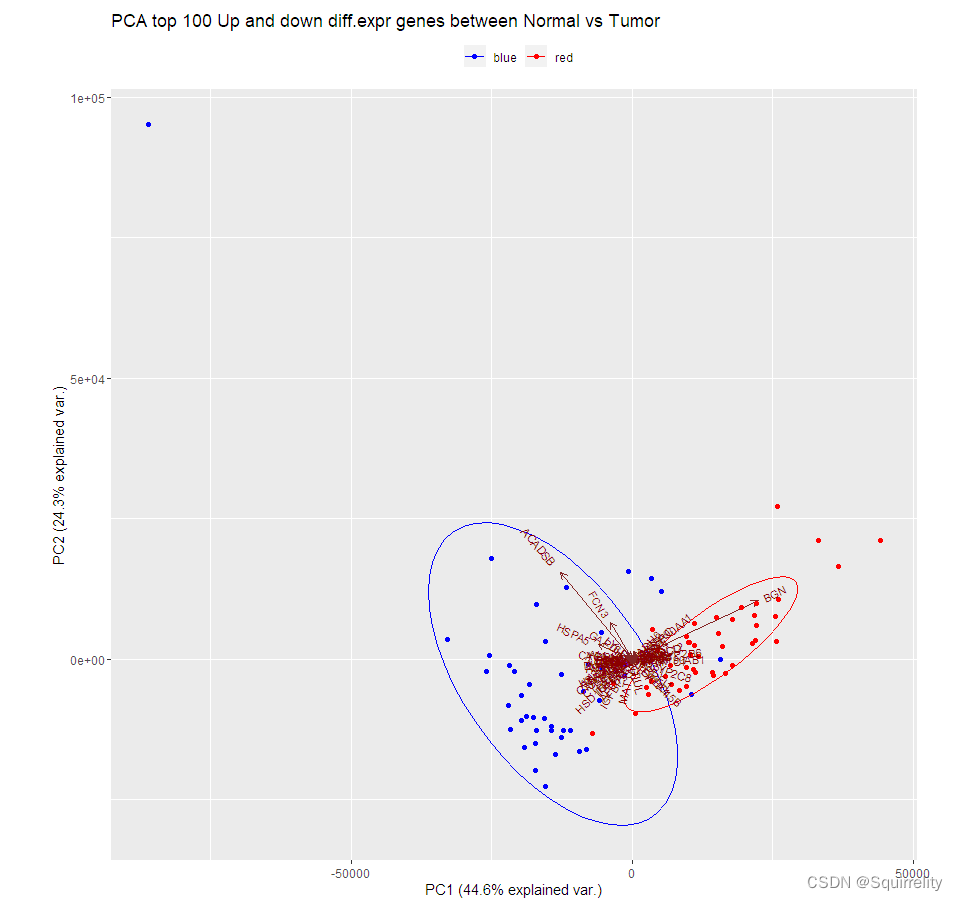
1.PCA主成分分析
TCGAvisualize_PCA()实现主成分分析的主要用法:
TCGAvisualize_PCA(dataFilt, dataDEGsFiltLevel, ntopgenes, group1, group2)

#标准化
dataNorm <- TCGAbiolinks::TCGAanalyze_Normalization(data2, geneInfo)#质量控制
dataFilt <- TCGAanalyze_Filtering(tabDF = dataNorm,method = "quantile", qnt.cut = 0.25)#选择正常样本
group1 <- TCGAquery_SampleTypes(colnames(dataFilt), typesample = c("NT"))
#选择癌症样本
group2 <- TCGAquery_SampleTypes(colnames(dataFilt), typesample = c("TP"))#Principal Component Analysis plot for ntop selected DEGs
pca.top200 <- TCGAvisualize_PCA(dataFilt,dataDEGsFiltLevel, ntopgenes = 200,group1, group2)
上面代码生成的图如下所示。
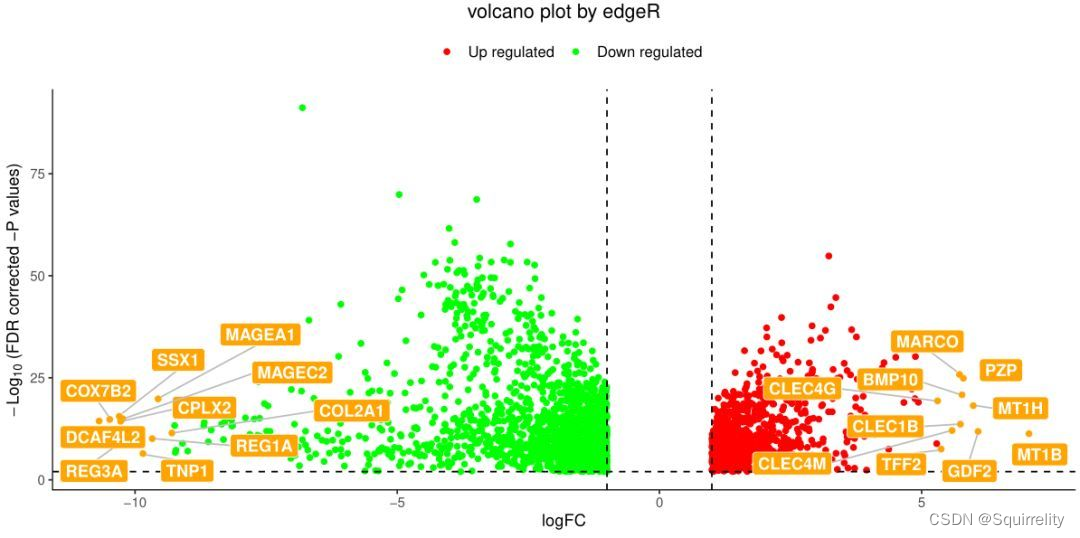
2.火山图
#为了做图的需要,突出显示logFC≥8的gene名称
DEG.BRCA.filt<-dataDEGs[which(abs(dataDEGs$logFC) >= 8), ]
str(DEG.BRCA.filt)
#'data.frame': 29 obs. of 5 variables:
#说明共有29个基因满足|logFC|≥8TCGAVisualize_volcano(dataDEGs$logFC, dataDEGs$FDR,filename = "TumorvsNormal_FC8.edgeR.pdf", xlab = "logFC",names = rownames(dataDEGs), show.names = "highlighted",x.cut = 1, y.cut = 0.01, highlight = rownames(dataDEGs)[which(abs(DEG.LIHC.edgeR$logFC) >= 8)],highlight.color = "orange",title = "volcano plot by edgeR")
上面代码生成的图如下所示。突出显示了logFC≥8的gene名称
3.GO功能分析条形图
TCGAbiolinks 输出条形图,其中包含三个本体的主要类别(分别为GO:生物过程、GO:细胞成分和GO:分子功能)的基因数量。
ansEA <- TCGAanalyze_EAcomplete(TFname = "DEA genes Normal Vs Tumor",RegulonList = dataDEGs$gene_name
) TCGAvisualize_EAbarplot(tf = rownames(ansEA$ResBP),GOBPTab = ansEA$ResBP,GOCCTab = ansEA$ResCC,GOMFTab = ansEA$ResMF,PathTab = ansEA$ResPat,nRGTab = dataDEGs$gene_name,nBar = 10
)
上面代码生成的图如下所示。