参考我的上一篇博客https://blog.csdn.net/weixin_62528784/article/details/142132891?spm=1001.2014.3001.5502,
在处理完hic上游分析模块之后,接下来就是正式的3层次模块分析了,compartment+TAD+loop 3大主层次,本篇介绍compartment层次。
1,compartment的识别(未涉及区室转换):
(1)使用homer:
参考https://mp.weixin.qq.com/s/qH4l6Igx9p1zSv4h-IW9Dg以及https://mp.weixin.qq.com/s/6FxPrvFSutIWQ0SrHrQ_Dg,当然这些都是简单的示例,目的是为了让我们清楚应该使用homer的什么工具函数,然后再去找英文教程,github上对应homer函数/脚本+关键词搜一大堆;
(2)使用GENOVA:推荐使用
相关指南参考https://github.com/robinweide/GENOVA/blob/master/vignettes/GENOVA.pdf,

上面是新的,下面是旧的。
①首先是数据导入:
GENOVA中load_contacts函数可以导入上游不同的hic数据格式,包括hic-pro原生输出格式、hic格式、cool格式/mcool等;

为了与我们前1篇博客相统一或者前几篇博客相统一,我们只认为hic-pro原生输出数据格式以及矫正之后的mcool格式比较稳妥可靠,hic格式不考虑,
只考虑这两种格式,可以到GENOVA中issue取经,参考经验。
现在假设我们所有的数据都由hic-pro原生输出转换到了mcool格式,现在如何将mcool格式数据导入到GENOVA中?
参数主要参考<https://github.com/MaybeBio/PDXHiC_supplemental/blob/main/Fig4_HiC_misc/05_ABsaddle.Rmd>中的load_contacts部分,

问题主要在于其中的balancing参数(其他的参数都没有太大问题,centromeres实际上可以不提供bed数据,因为default有默认的处理),

参考https://github.com/robinweide/GENOVA/issues/346,这个问题问的就是balance的问题:

所以应该使用balance = TRUE吗?
我看了一大圈issue中,大部分人使用的都是hic格式数据,但是大多数都是T,实际上这个也是default的设置。
那默认的设置就不要改?
建议进入这个代码中看一看:
https://github.com/robinweide/GENOVA/blob/master/man/load_contacts.Rd
进入到cool格式导入的函数中查看:
https://github.com/robinweide/GENOVA/blob/master/R/loading_cooler.R


其实看了一大堆,也还是不清楚;
再参考问题https://github.com/robinweide/GENOVA/issues/242,
balance与norm的关系是什么?

也不是很清楚。
总之,重申一下:
首先参考我们上一篇的博客,我们的hic-pro转换到cool,先是hicNormalize --normalize smallest再是hicCorrectMatrix --correctionMethod KR,然后合并为mcool,总之mcool文件经过了KR矫正!
官方参数:
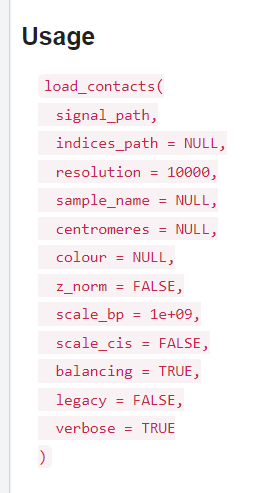
除了上面的balance问题,
还有就是scale_bp以及scale_cis参数,后者是false暂时不用考虑;但是前者怎么处理?
参考https://github.com/robinweide/GENOVA/issues/356,

提问者实际上也提出了同样的问题,他使用了h5到cool,也是KR矫正过了(我们是hic-pro到cool,也是KR矫正过了),
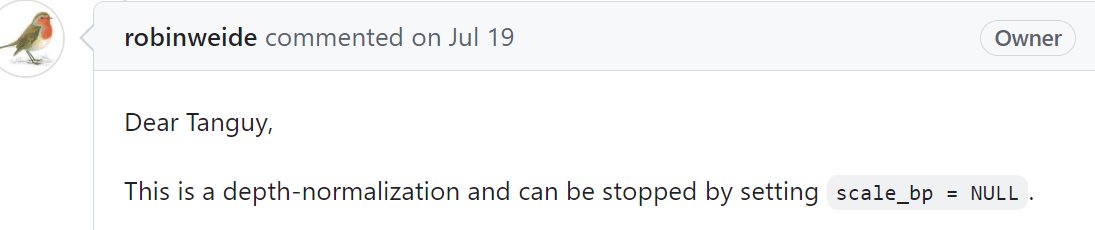
开发者建议使用scale_bp=NULL

综上:
我们的hic-pro转换到cool,先是hicNormalize --normalize smallest再是hicCorrectMatrix --correctionMethod KR,然后合并为mcool,总之mcool文件经过了KR矫正!
但是我们在load_contacts中有2个参数的设置感到困惑:scale_bp以及balancing!
干脆直接问开发者:待解决issuehttps://github.com/robinweide/GENOVA/issues/361
参考hicexplorer以及GENOVA中的issue来寻求经验,关键词:balance = TRUE以及load_contacts,
实在有问题就安装dev开发版本。
暂时按照balance=T以及其他设置都默认的方式导入到GENOVA。
②导入了数据,之后就是区室识别的方法了:
compartment_score函数,结果用visualise函数可视化(辅助ggplot2)——对区室分数PC1的±可视化分析,实际上就是AB区室识别;
2,saddle分析:
脚本参考修改后HiC_pipeline/pipeline/hic 基础pipeline骨架/01_ABsaddle.Rmd+PDXHiC_supplemental/Fig4_HiC_misc/05_ABsaddle.Rmd原代码
承接上面第1部分GENOVA中导入了mcool文件,
(1)compartment_score函数,结果用visualise函数可视化(辅助ggplot2)——对区室分数PC1的±可视化分析,实际上就是AB区室识别;


A/B compartments信息是由交互矩阵经过PCA降维得来,具体哪些是A哪些是B,需要明确一下,一般认为值>0为A成分,值<0为B成分;
signed compartment scores,就是计算特征向量;
performs an eigenvector decomposition of the observed / expected matrix minus one.

对相关系数矩阵进行PCA降维分析,在第一主成分PC1轴上,根据正负可以将染色质区域分为A/B compartment;
参考https://mp.weixin.qq.com/s/tFfxy0AfCWKg9novBddC6Q等,

就说是对互作矩阵进行OE归一化,计算P相关系数,对相关系数矩阵进行PCA降维分析
在PC1轴上,依据符号sign正负将染色质分为AB区室;
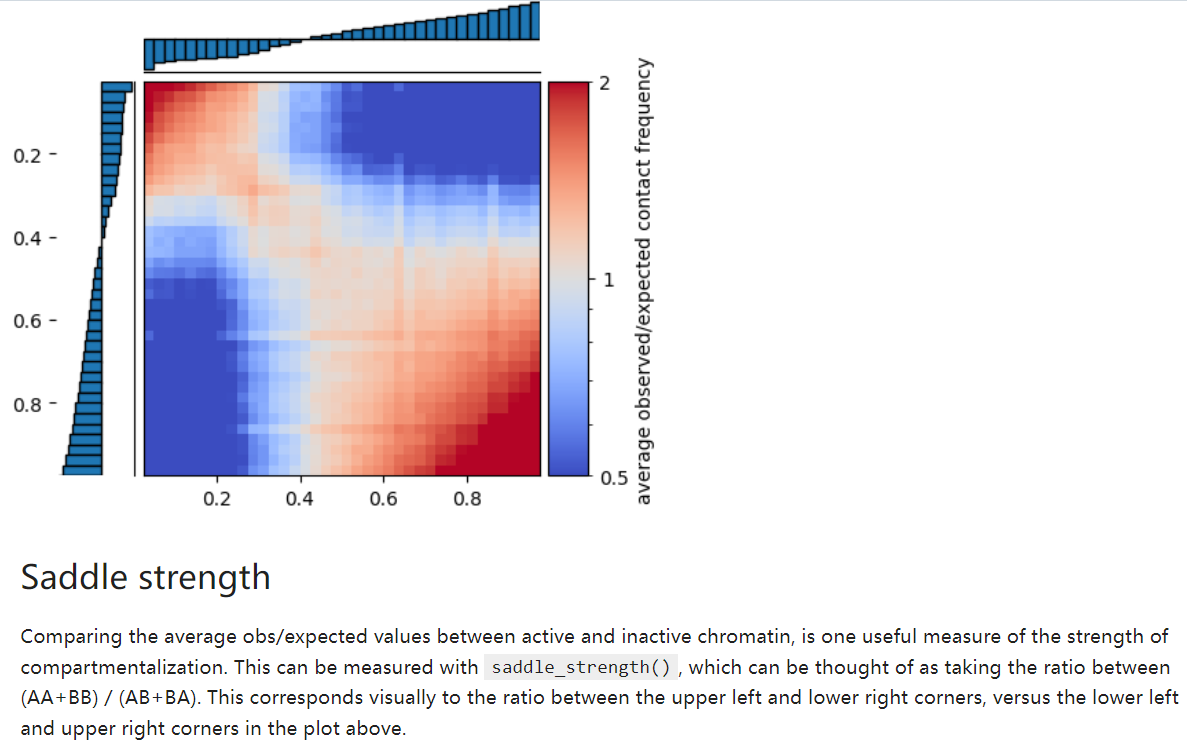
(2)对compartment_score的结果,使用saddle函数进行saddle分析,结果用quantify函数进行定量化,对quantify的统计结果提取数据用于分析Compartment-strength;
saddle有:1个是用GENOVA的saddle函数,另外1个是用其他脚本重新编写
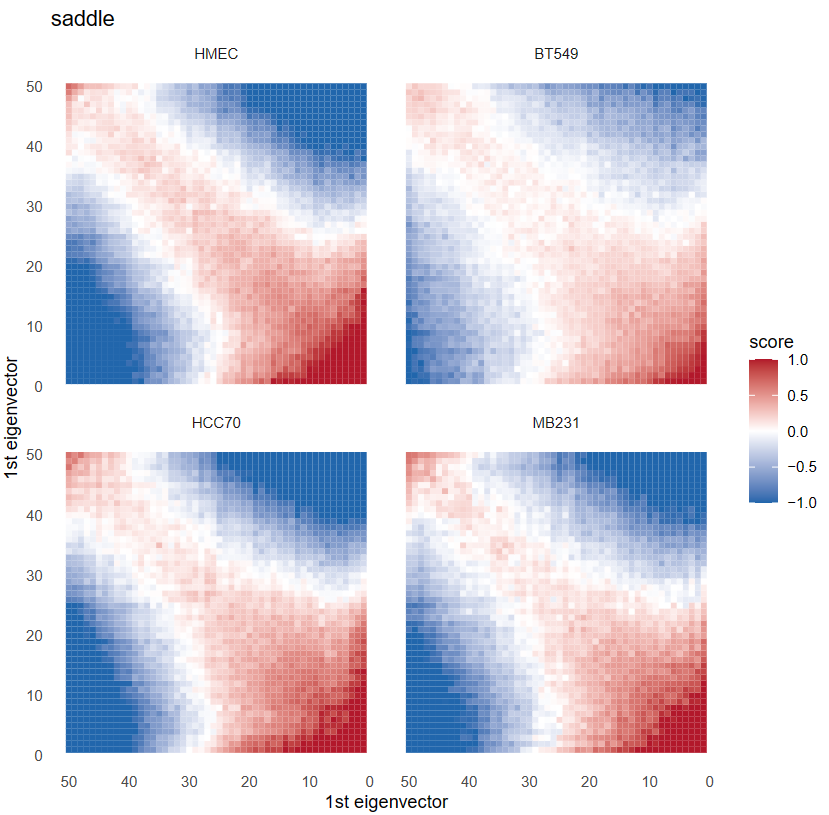
首先分析saddle之前肯定要calling AB区室,然后所谓的AB区室实际上是由交互矩阵经过PCA降维之后获得的1st特征向量EV(即PC1)的取值所定义的;
参考分析https://cooltools.readthedocs.io/en/latest/notebooks/compartments_and_saddles.html,
A/B compartments信息是由交互矩阵经过PCA降维得来,具体哪些是A哪些是B,需要明确一下,一般认为值>0为A成分,值<0为B成分;
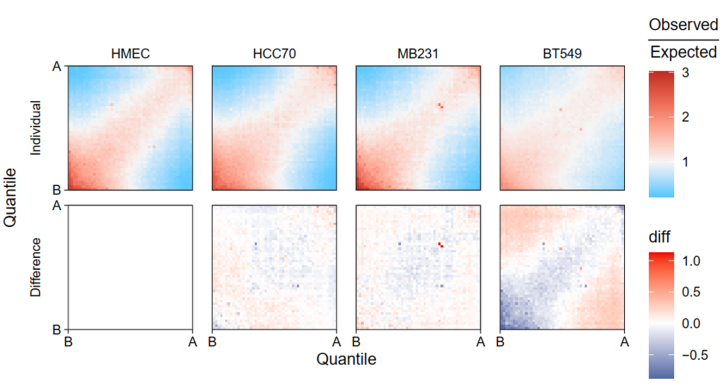
所以我觉得saddle图实际上就是对于AB区室的一种可视化,很直白地分析了EV1的分布,然后就是分析不同状态下特征向量所呈现出的偏好,根据其数字化特征向量PC1值分组的区域之间的平均观察/期望接触频率,知道AA、BB、AB、BA四种类型的交互情况,即交互数量和交互强度的估计,可谓一眼就能知道染色质的开放情况;
参考其中对于saddle的解读https://github.com/open2c/open2c_examples/blob/master/compartments_and_saddles.ipynb
中文资料相关鞍点图的只有:https://mp.weixin.qq.com/s/sDcm97GmK6-bD6rormRgAg
what:saddleplot作为展示A/B compartments的图,可以很轻松的知道AA、BB、AB、BA四种类型的交互情况,即交互数量和交互强度的估计,可谓一眼就能知道染色质的开放情况。如果多个条件的样本放在一起比较,便可以知道不同条件对染色质开放的影响。
展示染色质区室开放及交互情况的热图,
how:如何解读
Saddle分析是一种用于比较染色体亚区(compartment)之间相互作用的一种方法。以下是对Saddle分析内容的解释:
Compartment与Compartment分数:
Compartment指的是染色体上的亚区,这些亚区在空间上可能具有不同的结构和功能。
Compartment分数是对每个亚区进行的一种度量,用于表示该亚区在三维染色体空间中的局部结构或功能。
Compartment分数的分位数对比分析:
Saddle分析进行了分位数对比,将Compartment分数的分位数区间进行比较。
对每个染色体臂(chromosome arm),将Compartment分数分为多个分位数(quantile)区间。
计算观察到的与预期的接触比:
针对每一对分位数区间的组合,计算观察到的与预期的接触比。
首先,将该分位数区间中所有基因组成对的接触数进行平均。然后,将该分位数区间中所有基因组成对的接触数与与对角线相同距离处的平均接触数相比较,得到观察到的与预期的接触比。
观察到的与预期的接触比的计算:
观察到的与预期的接触比是通过将两个亚区之间的接触数与相同距离处对角线上的接触数进行比较得到的。
这种比值允许检测到染色体中的空间亚区之间的相互作用模式是否超过了随机期望的水平。
总之,Saddle分析提供了对染色体亚区之间相互作用模式的定量比较,通过比较不同Compartment分数分位数区间中的观察到的与预期的接触比,可以揭示染色体结构与功能的相关性。

所谓的鞍点saddle分析实际上就是chr内部的区域自我偏好分析,
依据compartment score将每条chr划分为50个分位数区间,分位数靠前的一般也是正的就是A,分位数末尾的一般也是负的就是B,
然后就是组合分析,50个bin之间所有组合都分析gene互作频率域预期互作频率的比值,
说白了确实是分析bin之间的互作偏好,
那热图实际上只能查看互作强度,互作数量其实是可以通过其他条形图之类分析的,
最后每条chr上的互作情况平均之后就是整体chr。

让我们再来总结一下:


参考saddle分析的原理https://blog.sciencenet.cn/blog-2970729-1367103.html,
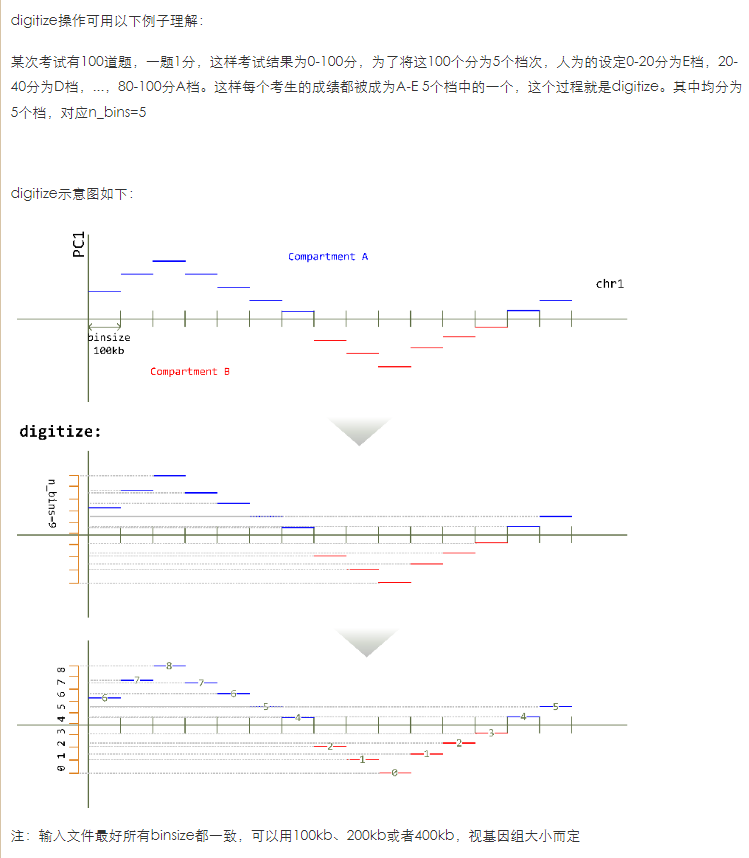
就是依据PC1值将区室区域chr分为nbin,我的代码中是分为50bins/区域,
这里是依据PC1的值将所有区域分为50个分位数区间,不是简单将chr区域均分为50个区间;
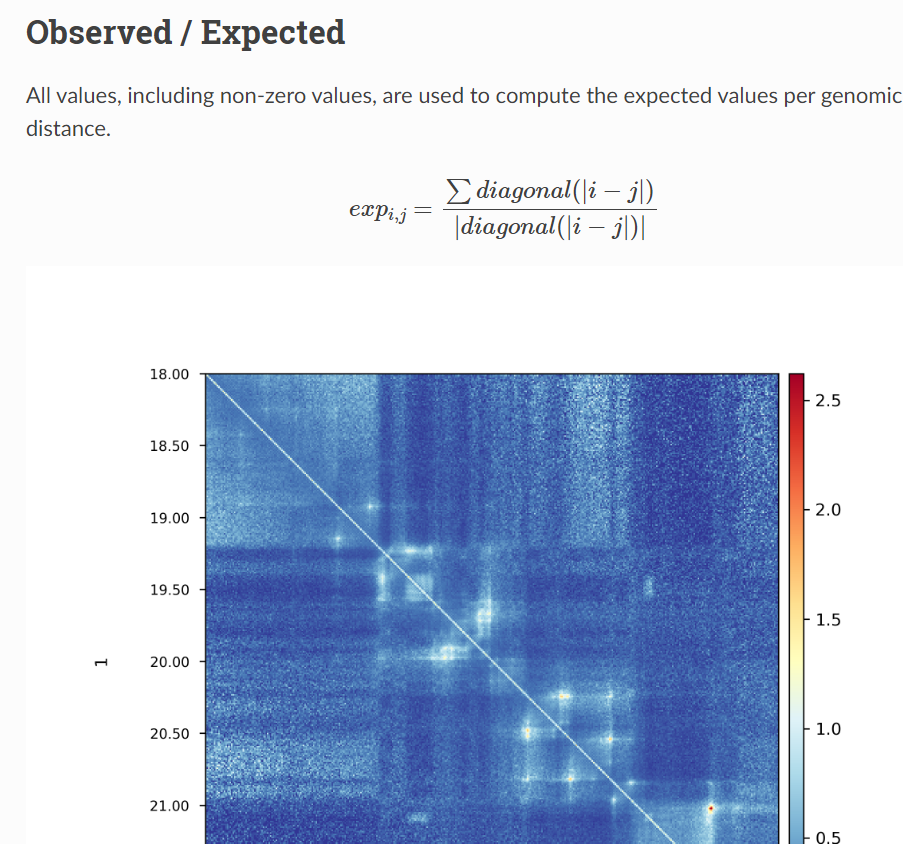
另外OE矩阵也可以从这里理解:
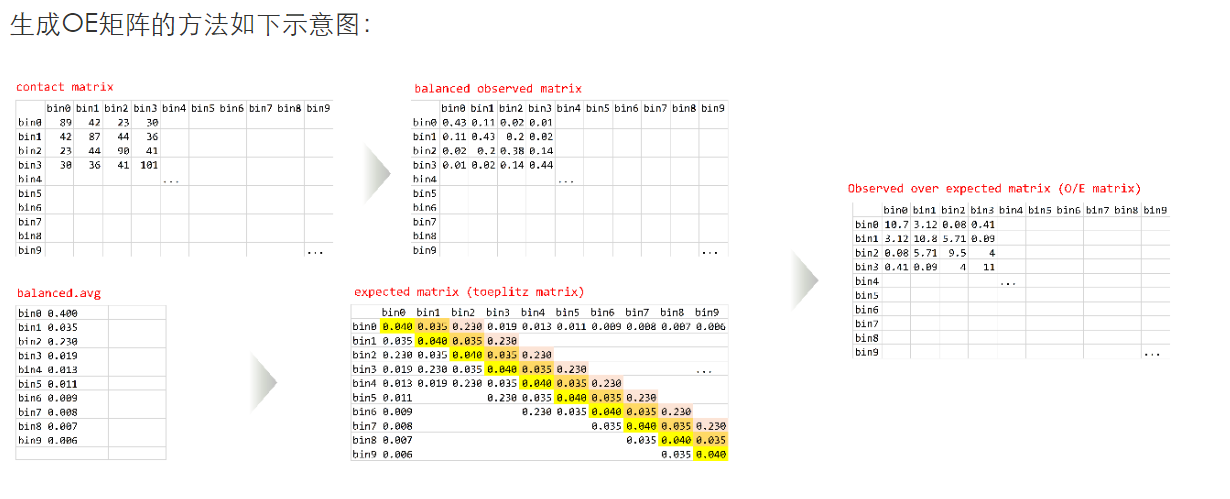
然后就是如何理解OE矩阵:
https://hicexplorer.readthedocs.io/en/latest/content/tools/hicTransform.html
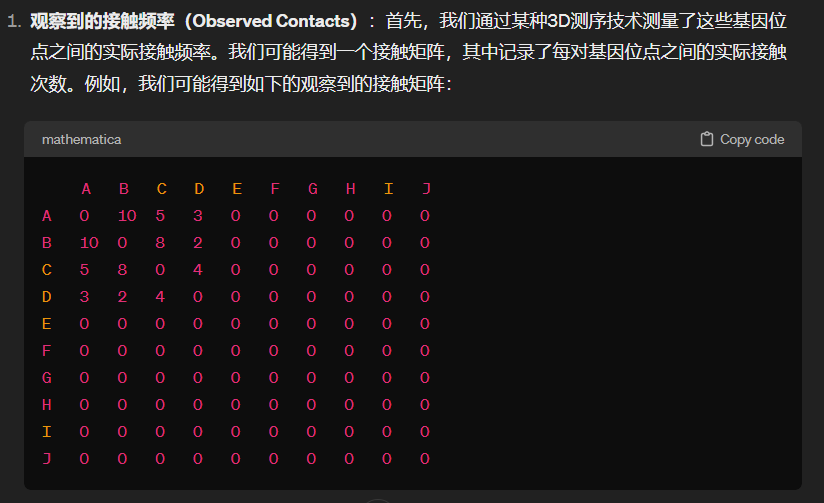
大概意思是,首先原来的互作矩阵,应该是ij区域对应互作回帖的reads数目作为contacts(PE测序然后回帖回去定区域i与j,然后reads数目作为矩阵内值),
然后矩阵会经过balance(ICE,KR之类),这是observerd的矩阵
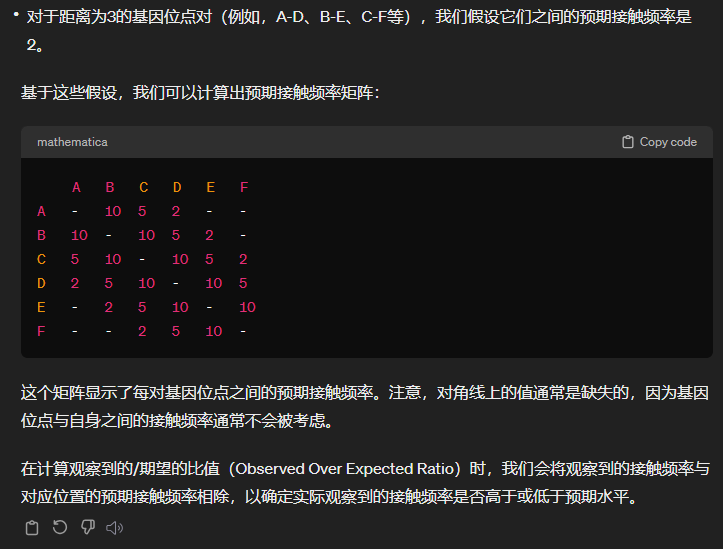
然后expected的矩阵就是:如果选取的区域是ij,那么就计算所有距离为i-j绝对值的那些配对区域对,然后同样是计算reads作为contact之类,然后将所有这些区域之间取个均值,就是所有i-j类型的expected,
然后O/E就是结果;



GENOVA中的设置就是contact/对角线上相同距离average,
效果可以优化:交互数目看红蓝色块bin的数目多少,强度看颜色

#这是一条分割线
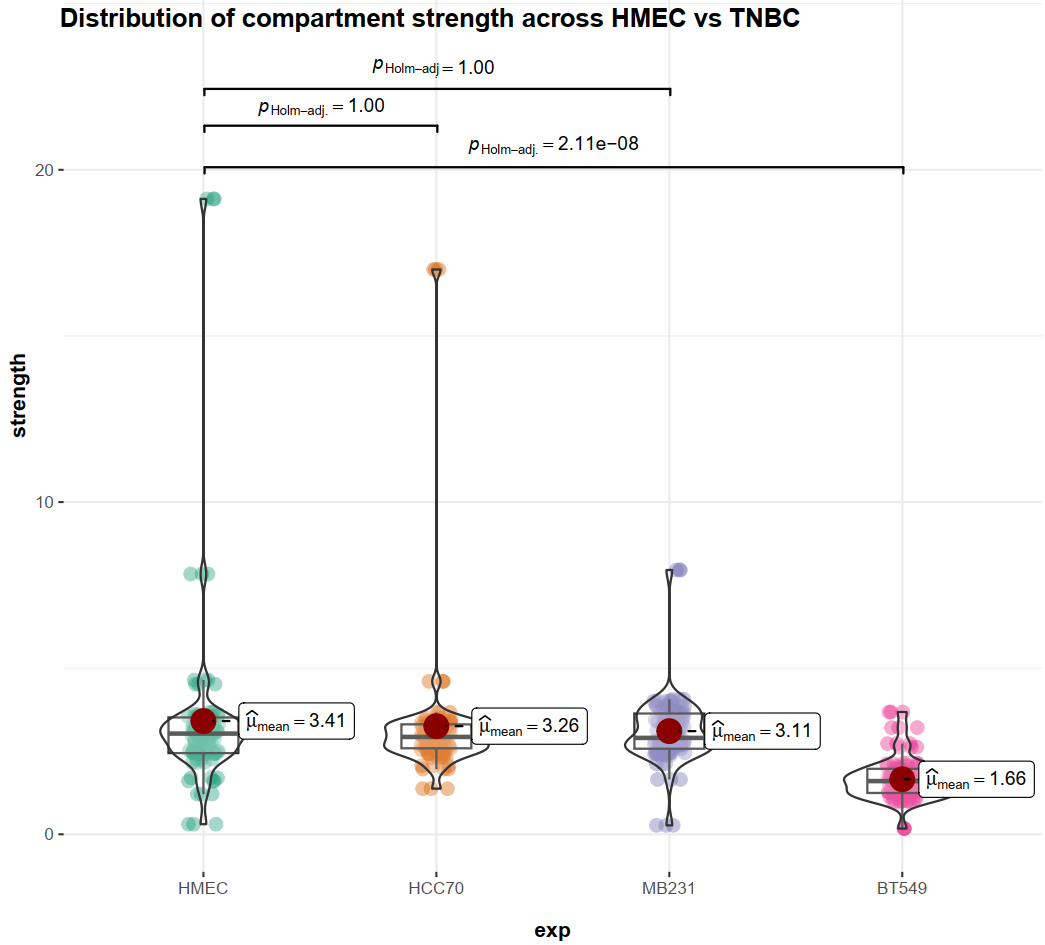
Compartment-strength有:
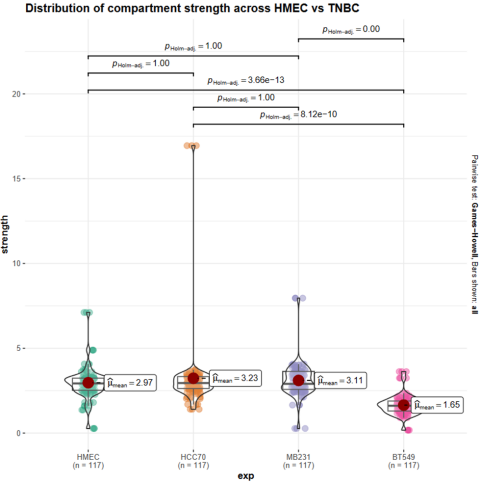

是对AA+BB互作比对AB互作的一种评分:
measure of how well A and B compartments are separated from oneanother
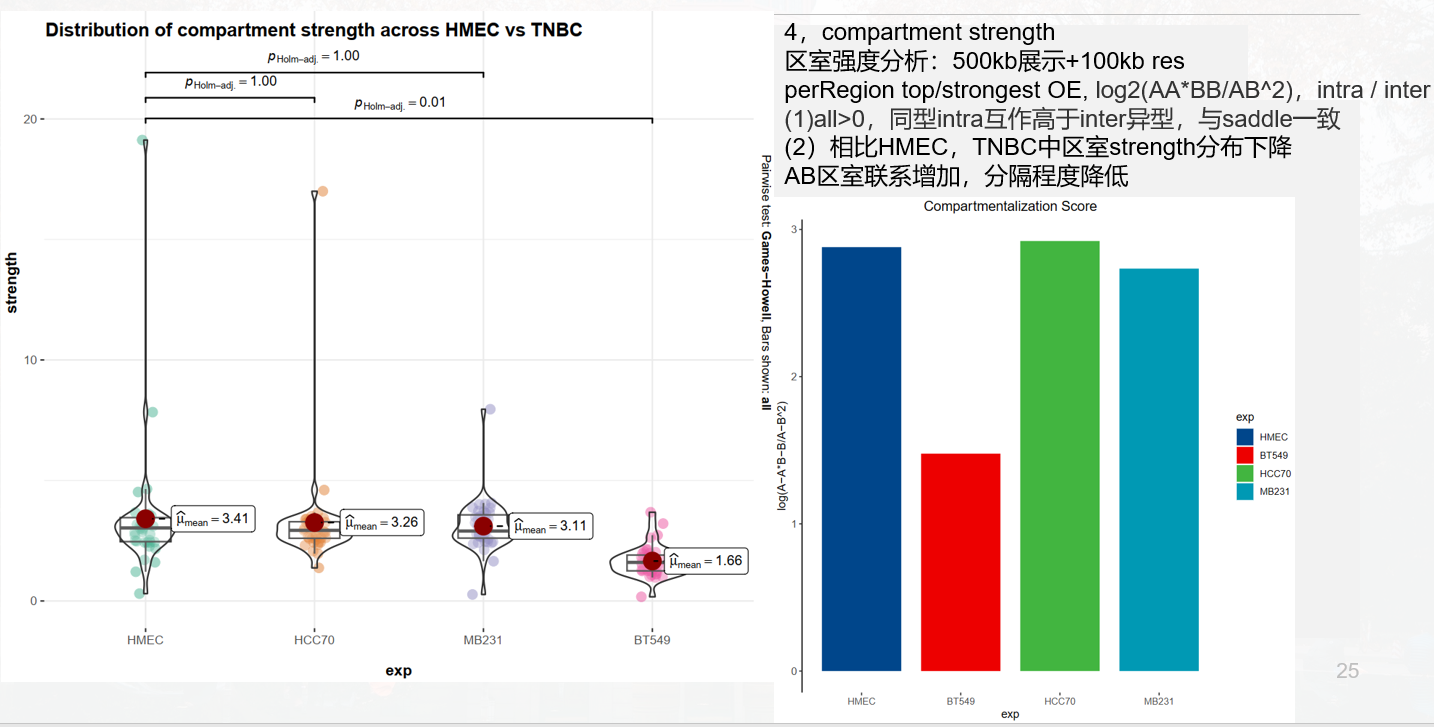
所以值越高是区室AB联系越不紧密,即分隔程度;
所以HMEC普遍区室强度较低,也就是AB区室联系增强;



建议参考GENOVA原文献https://academic.oup.com/nargab/article/3/2/lqab040/6281451,
分数为1意味着室内相互作用与室间相互作用一样普遍,而较高的分数意味着室内相互作用更普遍,
所以就是将A-A+B-B区室内互作/A-B区室间互作,实际上还是saddle互作偏好的分析;
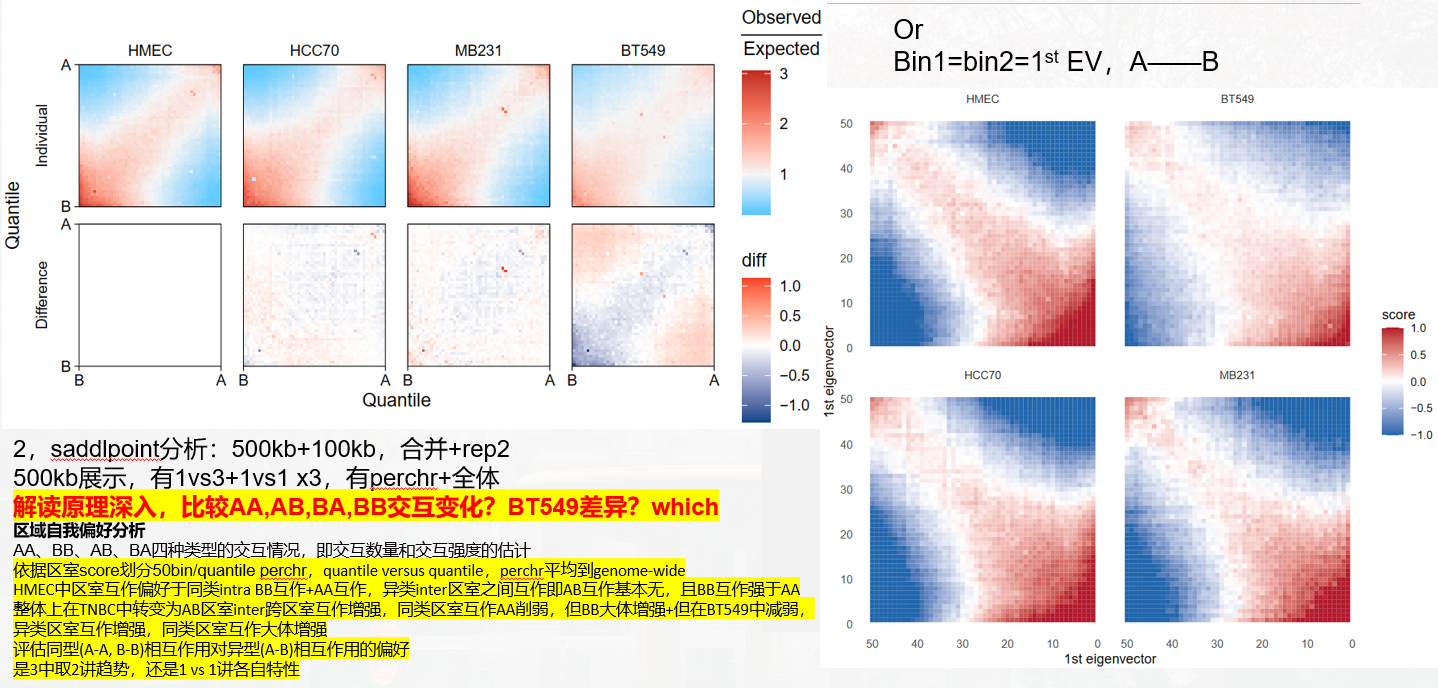
区室强度chr比较:具体意义参考GENOVA,离散3图
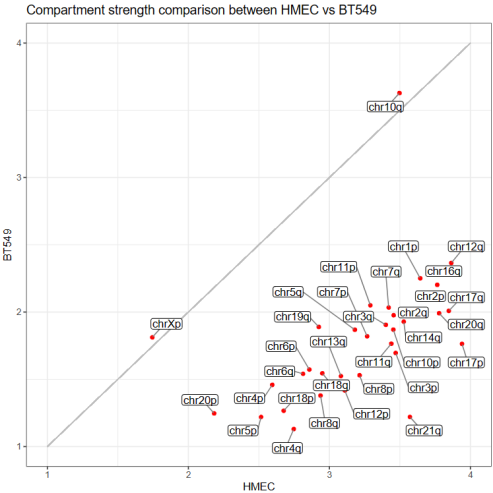
compartment strength differences,PQ长臂短臂,差异区域分析
对角线上的区域,TNBC区室score比HMEC高,即室内互作更显著更强+比例更高的区域,对角线之下的区域即室内互作互作比例更低的区域,值是看哪个互作为主+对角线是看哪个互作相比变化大;
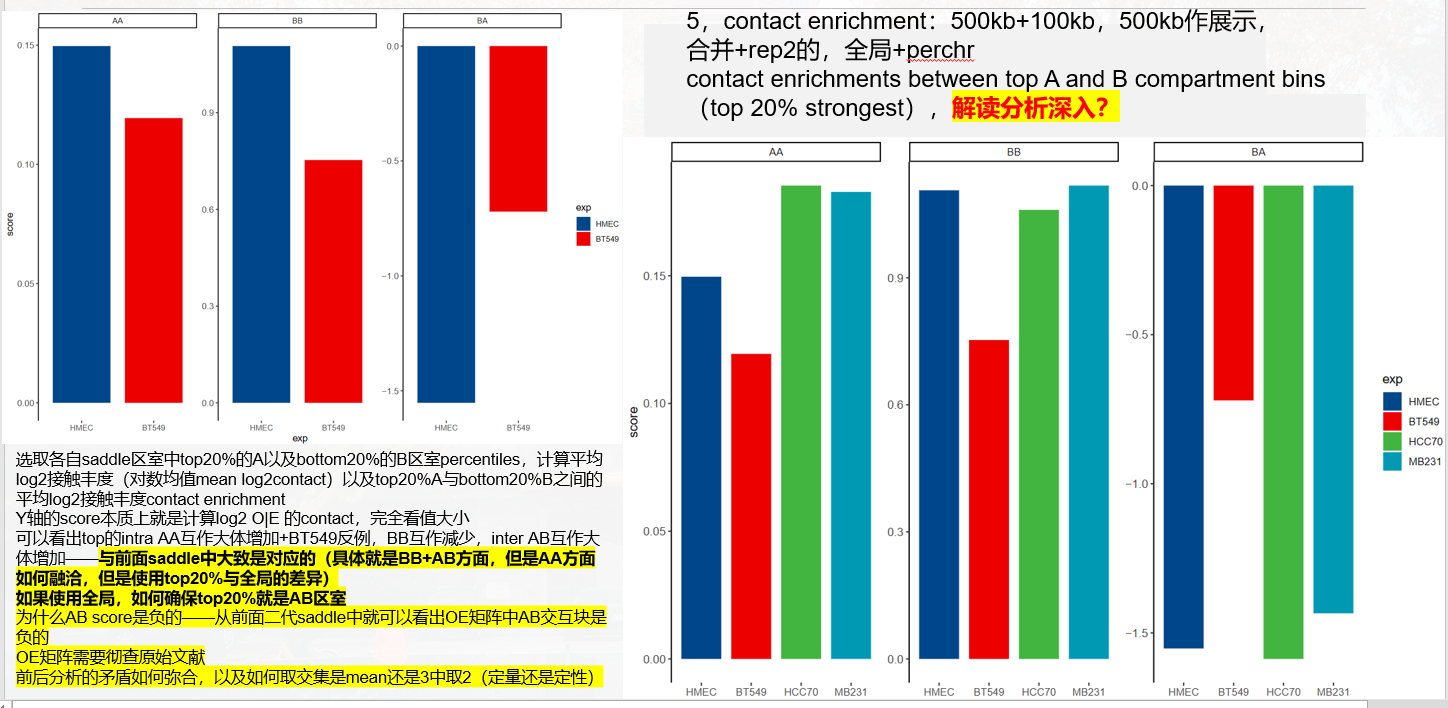
(3)对saddle中选取首尾20%的分位数区间nbins,分析Contact enrichment,实际上是将GENOVA中saddle的脚步进行重写,但问题是,怎么解释Contact enrichment这个变量的bio意义?——》待处理
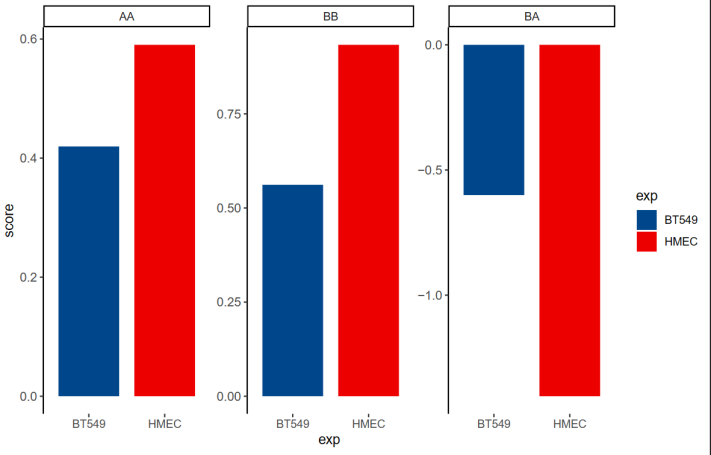
目前需要解读这里的contact enrichment,如何联系前后;
计算了 A-A 和 B-B 区室的摘要强度,这是在 PC1 百分位的顶部(A-A)或底部(B-B)20% 之间的平均 log2 接触丰度,也就是说AA,BB区室强度是选取top 20%或者bottom 20%的pc1百分位计算的contact(这里其实应该意识到按照pc1百分位分组的话前面肯定是A,后面肯定是B;只不过选取的是前面的A以及后面的B),所以是抽出了top的A以及bottom的B,分析互作score如何,比如说top的AA互作在BT549中比HMEC弱等等;
对于 A-B 区室强度,则是在 PC1 百分位的顶部和底部 20% 之间的平均 log2 接触丰度
使用 GENOVA v.1.0.0 R 包对 100 kb 分辨率的数据进行了迭代校正。
使用第一个特征向量(PC1)的值对 100 kb 的区间进行了分组,将其分为 50 个百分位组。
AB 区室分析:
在 50 个百分位组的两两组合中,计算了区间之间的平均接触丰度(obs/exp)。
将接触丰度的 log2 值绘制为热图,形成了 saddle plot(马鞍图)。
区室强度摘要:
计算了 A-A 和 B-B 区室的摘要强度,这是在 PC1 百分位的顶部(A-A)或底部(B-B)20% 之间的平均 log2 接触丰度。
对于 A-B 区室强度,则是在 PC1 百分位的顶部和底部 20% 之间的平均 log2 接触丰度。
选取各自区室中top20%的A以及bottom20%的B区室percentiles,计算平均log2接触丰度(对数均值)以及top20%A与bottom20%B之间的平均log2接触丰contact enrichment;
Why选取20%,能够确保对应的bins/percentiles是AB吗?(需要查看EV分布之类)
Y轴的score本质上就是计算log2 O|E的contact,完全看值大小,
可以看出top的intra AA互作大体增加,BB互作大体减少,inter AB互作 大体增加,相互比较好说,
为什么AB score是负的;
OE矩阵需要彻查原始文献,
前后分析的矛盾如何弥合,以及如何取交集是mean还是3中取2
总之y轴如何解读,表征的是什么,以及相互比较之下如何解读,如何和前面矛盾相互弥合,
为什么inter AB是负的,为什么是取20% top,
所以对于OE矩阵的解读以及算法实际中所涉及到的问题,都要回归到文献上去,
(4)Compartmentalization score分析:本质上也是选取首尾20%分位数区间,计算(AA+BB) / (AB+BA)相关比例
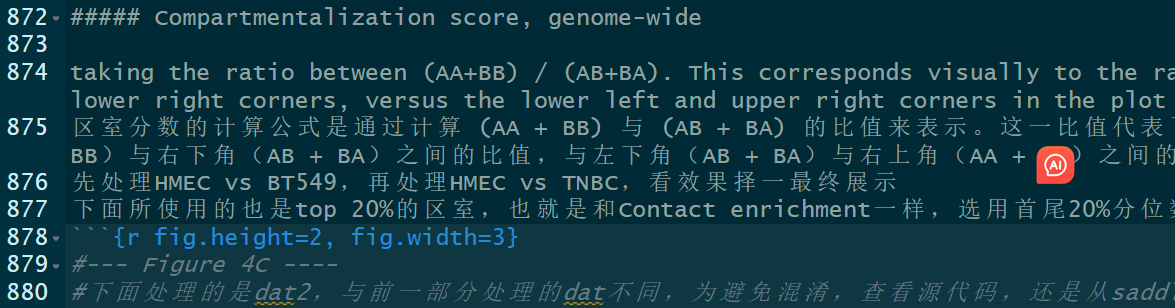
区室化分数分析

主要是解读什么是区室化分数,以及这个公式计算的是什么意义
taking the ratio between (AA+BB) / (AB+BA). This corresponds visually to the ratio between the upper left and lower right corners, versus the lower left and upper right corners in the plot above.
区室分数的计算公式是通过计算 (AA + BB) 与 (AB + BA) 的比值来表示。这一比值代表了热图中左上角(AA + BB)与右下角(AB + BA)之间的比值,与左下角(AB + BA)与右上角(AA + BB)之间的比值相对应;
综上:
可以参考PDX文献method中对saddle分析相关的3个概念:
heatmap saddle plot:Within pairwise combinations of the 50 percentile groups, average contact enrichments (obs/exp) between bins were calculated, and log2 of the contact enrichment scores were plotted as a heatmap saddle plot.
compartment strengths:Summarized A-A and B-B compartment strengths were calculated as the mean log2 contact enrichment between the top (A-A) or bottom (B-B) 20% of PC1 percentiles and between the top and bottom 20% of PC1 percentiles for A-B compartment strength, excluding chrY and chrM.
compartmentalization score:calculated as previously described using mean contact enrichments for A-A, B-B, and A-B following this formula
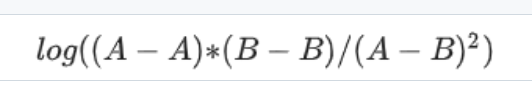
以下参考01_ABsaddle.Rmd脚本理解,尤其是自己新增部分,主要是经历絮絮叨叨
**(5)让我们再来总结一下:**以下是项目分析经验

下面的数目应该是:
23对总共24类chr,每条chr都有pq,然后每条chr上的pq上都有3类互作(AB,BA都是一样),
但是有些区域没有互作(可能是50个bin分类之后没有对应的交互值,所以为NA之后就没了,或者是共同影响的),总之范围是在24x3~24(类chr)x2(pq)x3(3类互作),有些chr上pq短臂可能没有互作3类所以略去了。
实际44x3左右,
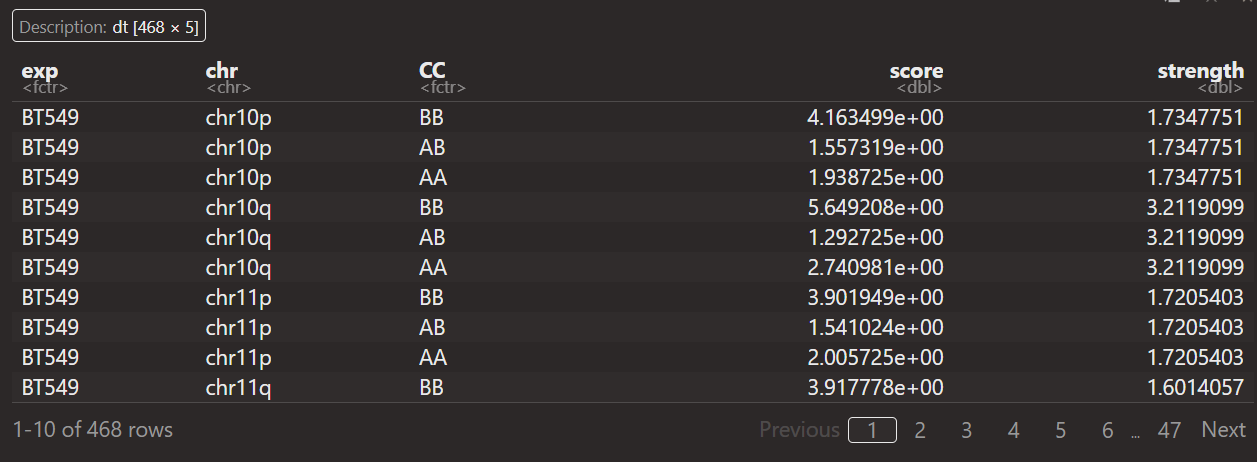
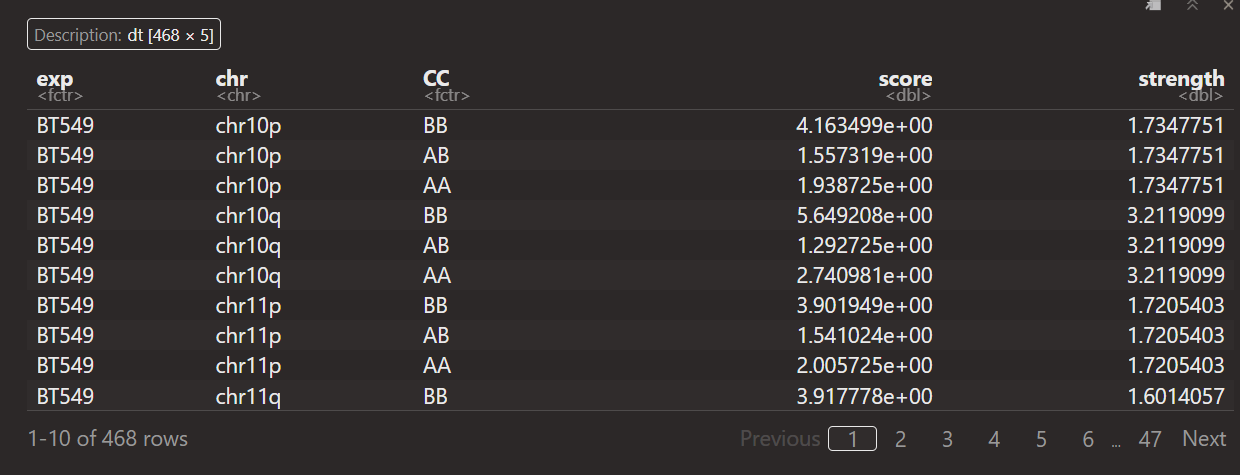
重点在于如何解释这里的score以及strength:
首先数据分析都是从saddle+quantity之后开始获得,然后score+strength数据对应的都是
每一条chr上pq区域等AA,BB,AB类型
那问题实际上就是对于某个chr上的某个短臂pq区域,
如何计算出cc以及score,strength这几列数据
算了,直接就说是汇总每条chr上区域AB信息(就不说AB的分位数了,这里就直接说是获取了每条chr上的AB区室信息),汇总AA,BB,AB区室间互作
intra AA+BB,inter AB区室互作等,这个score应该是OE值,这个还是可以理解的
但是strength是什么?其实可以发现
其实可以发现,每一条chr,无论是否有pq,CC都只有3种类型,就是intra AA,BB,AB
然后strength的话同一个区域是一样的(同一个p区域内,同一个q区域内)
所以strength本质上就是计算不同chr区域上的区室强度
就说是获得了不同chr区域上3类区室互作的OE值,以及区域本身的strength值
strength=在score上log2(intra AA*BB / inter AB^2)
可以看到实际上的strength值除了是intra/inter之外,还经过了log2转换,之所以这么做肯定是因为数据范围太大,离群值太多
可以看到strength差异确实大,而且这还是原算法中经过了log2转换的
可以手工排除异常区域:

源代码也是设定要排除的,只不过是排除整条异常chr区域,不是单单排除单个record,
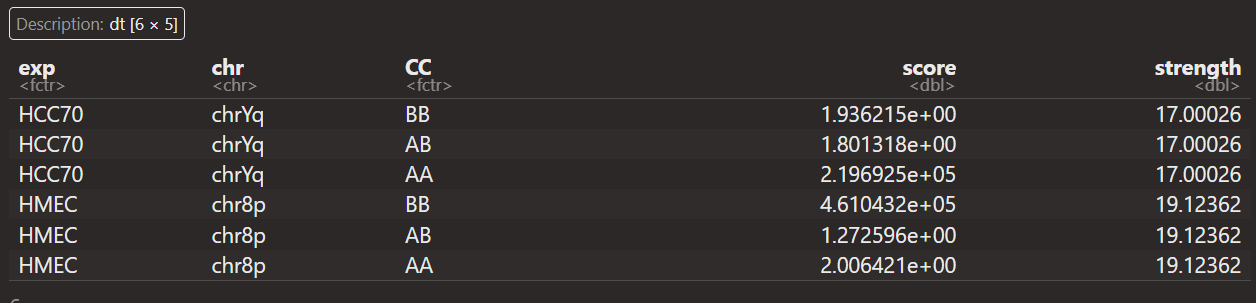
strength还是不排除异常值了。
然后有个问题就是如何获取整条chr上的AA以及BB以及AB互作的消息,我们前面是将整条chr的区域分成了50份分位数区间,如何从50个分位数区间的互作数据中获取整个区域的AA,BB以及AB互作的OE score,
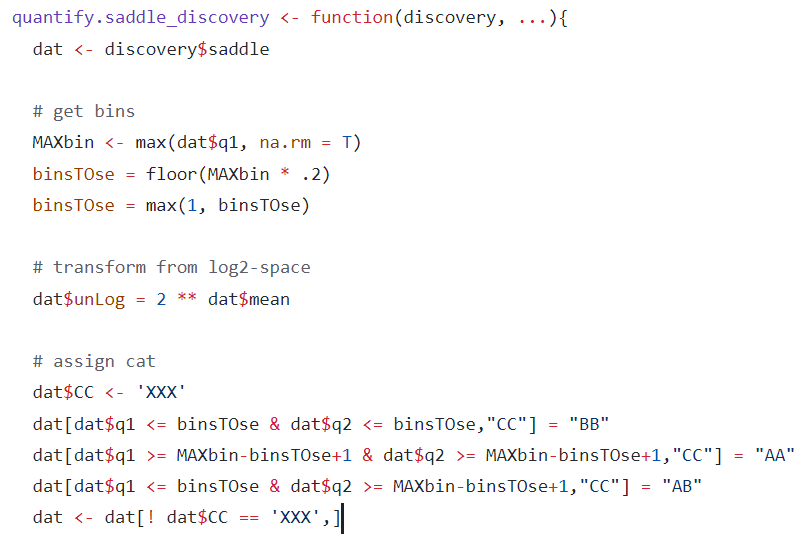
按照上面的算法,实际上使用的也是top20的bin,
所以整个思路就是:
saddle中是将整条chr区域划依据PC1值分为50个分位数区间(实际上是分短臂pq),然后计算50x50个分位数区间组合之间的OE值,来评估区室互作的偏好(交互的强度以及数目),然后又以top20的bin区间,即top20%的A作为A,bottom20%的B作为B,上下分界;以此为基础,获取AA,BB,AB的OE值,即最后定量化出来的score,然后获得了不同chr区域上3类区室互作的OE值,以及区域本身的strength值,strength=在score上log2(intra AA*BB / inter AB^2) ,所以实际上这里的区室strength值与后面的应该是一致的。
那么对于此处的数据,可视化的手段
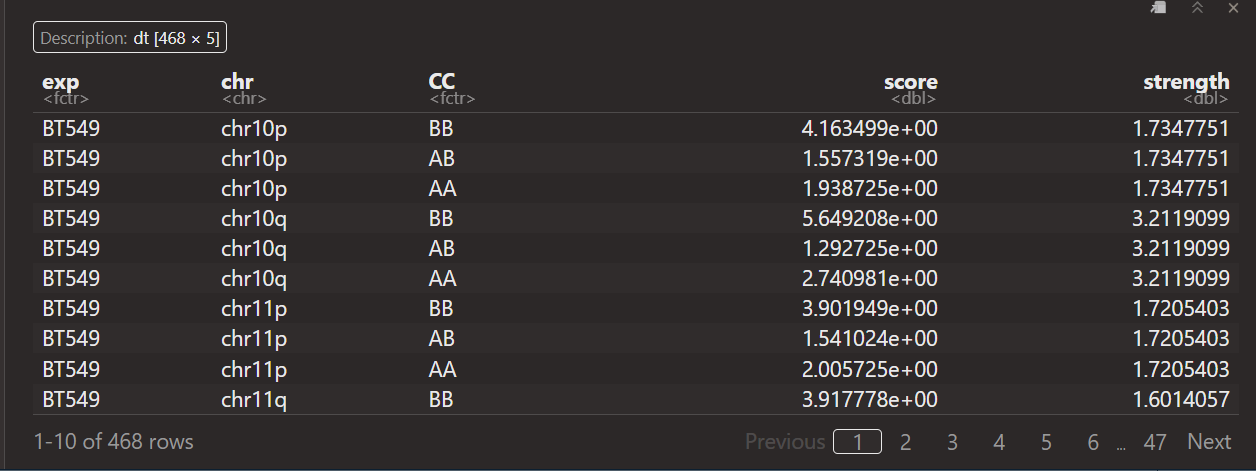
首先是perregion(实际上就是perchr中分pq)可以可视化3类区室互作的score,
然后就是strength本身是以region为单位统一的,所以可以按照region进行比较,
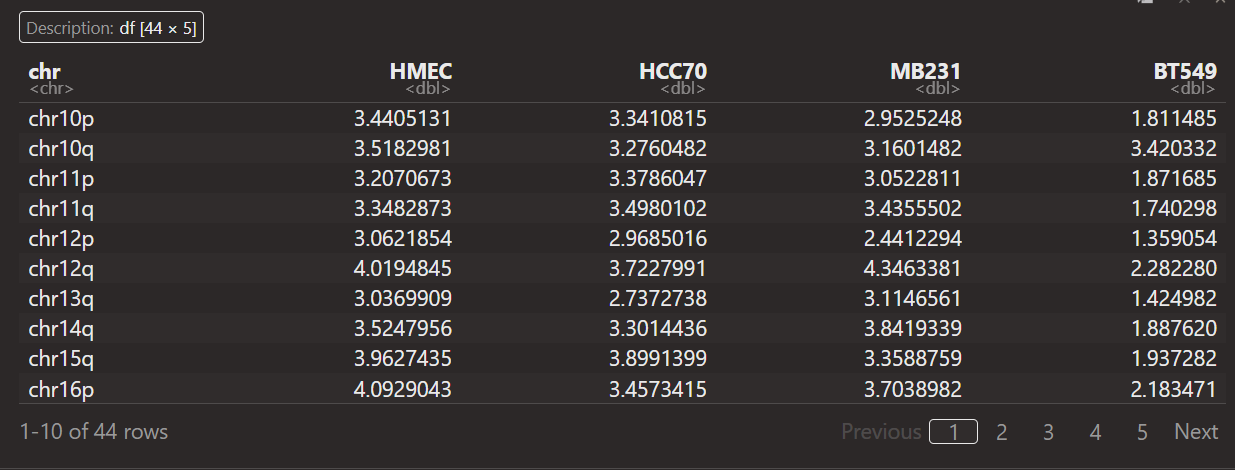
原始的css以及compared:
按理来说:
绘制box+vilion的图使用的数据就是这里的CSS的strength,但是如果直接使用这里CSS的原始数据的话,可以发现实际上是有重复的,因为3类区室都是一致的,每个strength值会有3个重复,
所以在脚本中才会提取出compared,使用unique,

也就是说:
strength可视化:使用compared或自己提取
score可视化:使用CSS
最后去除重复之后的数据:

展示的时候就这么说:
然后计算了每条chr区域上(top/strongest)3类区室互作的OE值,以AA*BB/AB^2来表征区室强度strength,并进行了log2转化,
如果原来的值大于1表明intra互作强于inter,有所偏好,log2之后就是大于0,perRegion OE
然后这一块内容和后面的区室整体化score实际上是一致的,所以也合并在一起;

但是同样的,放在一起效果不配套;
一个是看算法文档,是score的2指数化之后再mean再一个比值的log2,实际上比值就无所谓量纲了,另外一个是在OE值基础上进行的比值的log2,所以实际上是不一样的,也就是说我的箱线图不是OE的比值,而实际上是OE的指数化的比值,然后下面的才是OE层面的比值;

因为数据要配套,所以我同样可以和(5)score一样使用原来的数据进行处理
但是这里相反,因为箱线图的数据实际上是不能更改的,所以这里采用箱线图的数据,但是不使用OE score原始值,因为是比值,所以无所谓了,只要两幅图配合在一起即可
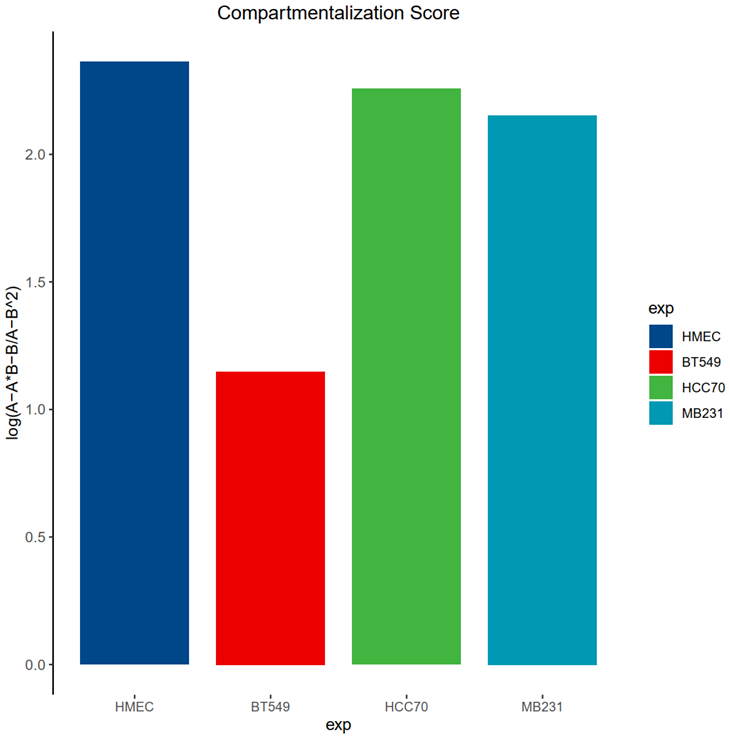
还是用了这个思路,但是算出来却值对不上?到时候就说是其他的计算方法
(或者这么说,一个是用工具函数计算出来的,一个是我用原始的OE score值计算的比值,但是在趋势上都是一致的)
至于compartment strength差异:
其实就是前面计算的perRegion的区室strength比较,每个区域的strength在3对比对中比较,
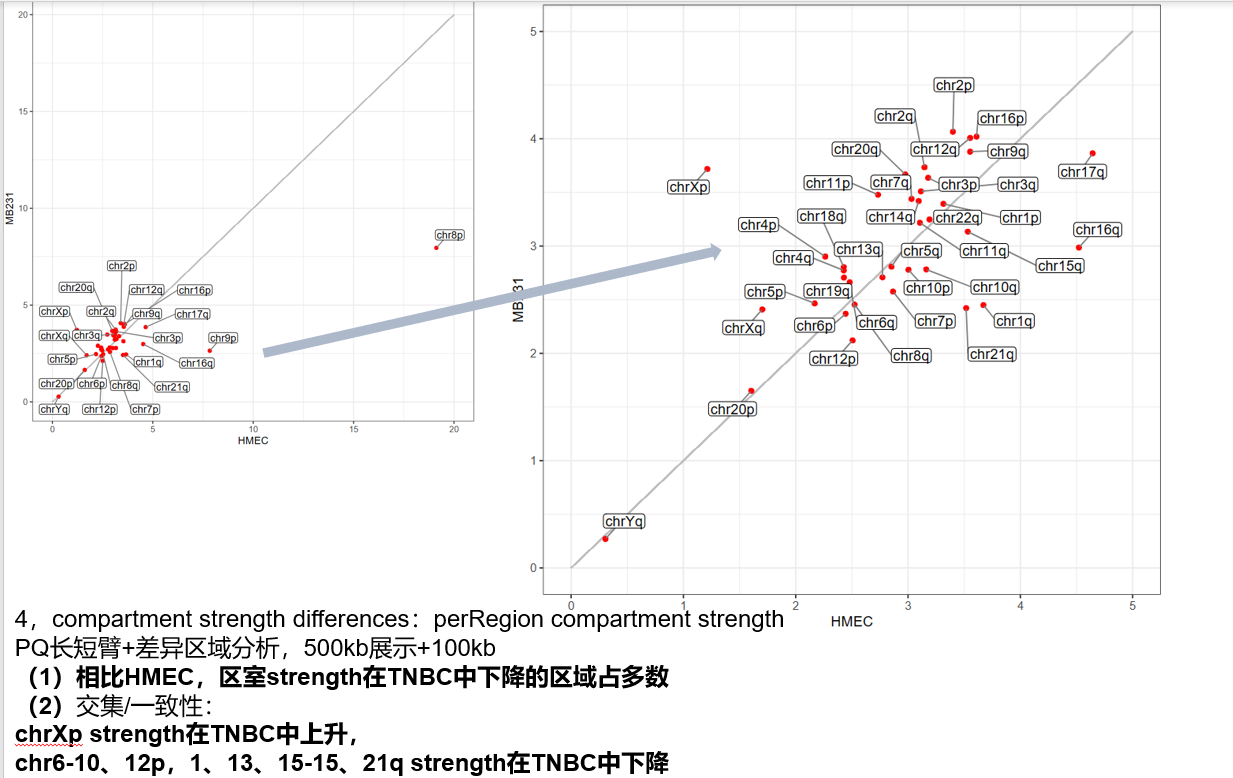
所以strength的可视化:
perRegion strength~样本,即(3)中saddle定量化
考虑区域pq,即如上
所以strength可视化分析的方面已经全部结束
实际上就是上面那副小提琴图,而且注意到GENOVA中实际上就是按照log2处理的,
所以对于异常值方面,只能人工排除;
至于compartment score方面:
strength可视化:使用compared或自己提取
score可视化:使用CSS
上面strength可视化的信息基本上已经完全结束,但是score方面
在原始的CSS数据中,还有每一个区域对应AA,BB,AB3类型的OE score值
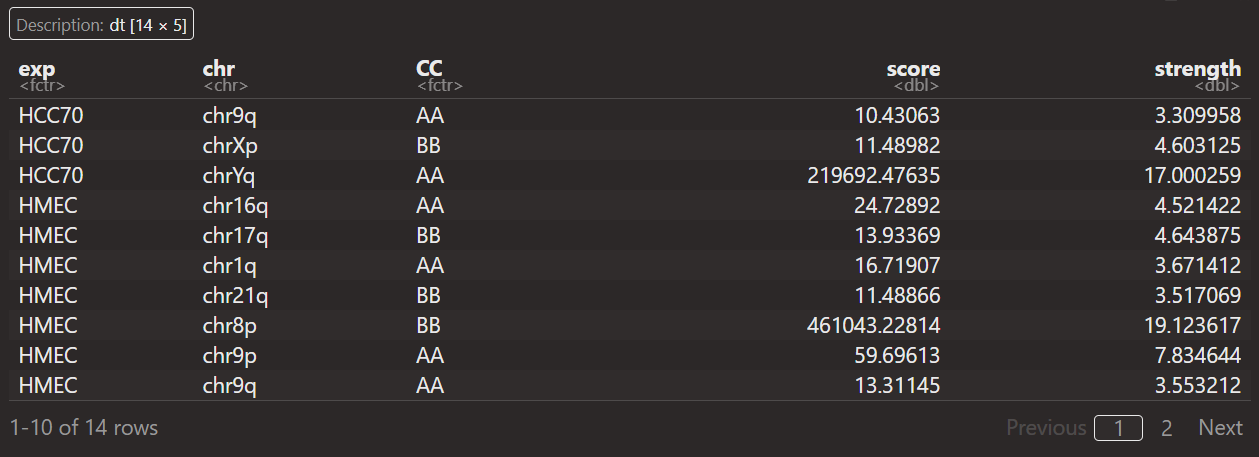
SCORE的可视化要expcc分组下可视化score,或者ccexp分组下可视化score,最好有个p检验,箱线图+小提琴图,至少得2个分组的箱琴图;
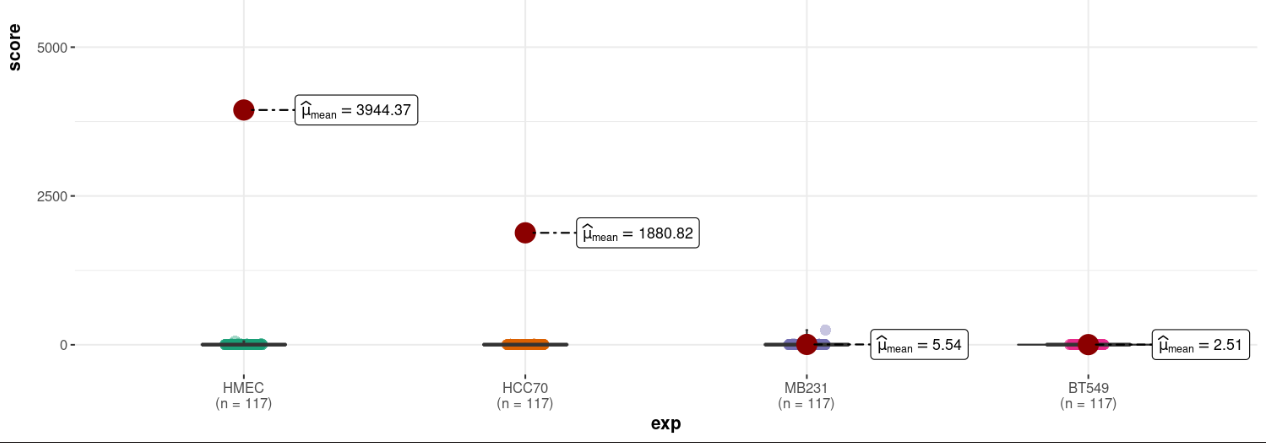
但是发现数据太过离谱,范围不一致,主要是AA以及BB中的异常值

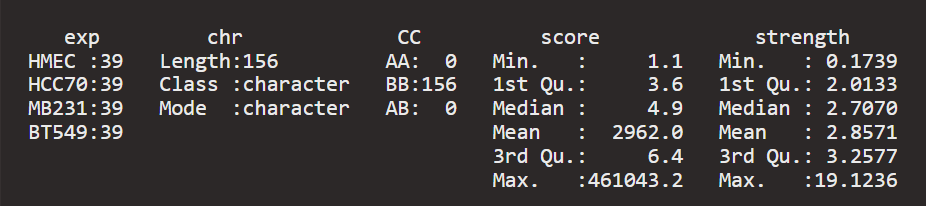

异常值比stength还离谱,必须进行排查以及矫正,查看异常值,进行排查
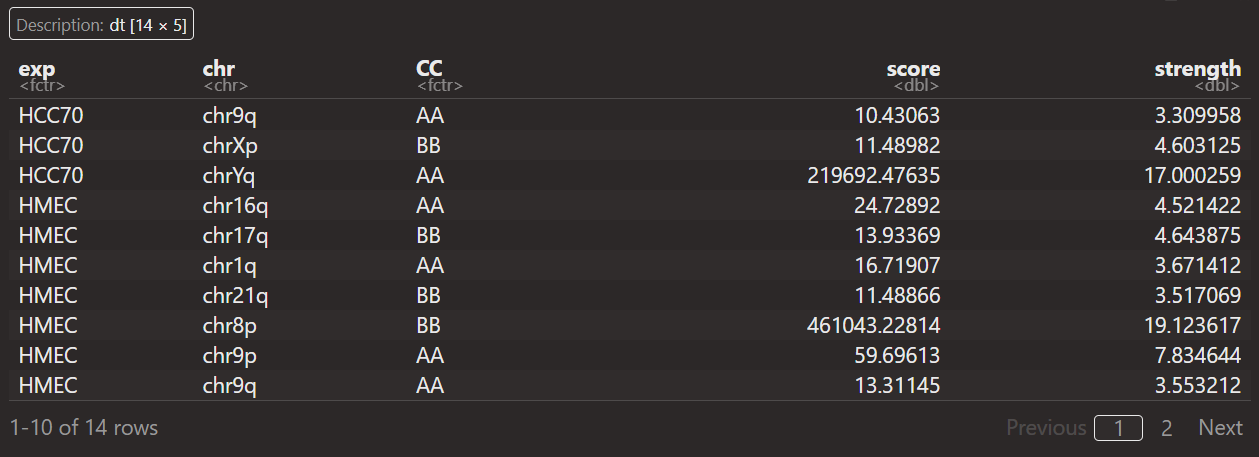

实际上可以发现Yq,8p都很异常,其实源代码脚本中也打算进行排除异常值

但是太一刀切了,可以只过滤掉部分对应sample的数据进行分析
可以如下处理:
①同样log2处理
当然如何处理数据都可以,主要是与自己已经有的结论相互符合,
主要是后面有一个计算contact enrichment的,
然后同样使用的是了类似于score的数据,然后也是取top以及bottom的20%
但是分析处理上两者是不同的:

我所处理的原始数据就是score,
然后我是将score log2转换,实际上就是将数据转换为原来的mean值,也就是saddle中最原始的bin vs bin,也就是上面中的unlog前的mean
然后脚本中处理那部分的数据是在mean上log2,所以实际上是最原始的数据的log2,也是我箱琴图中数据的log2
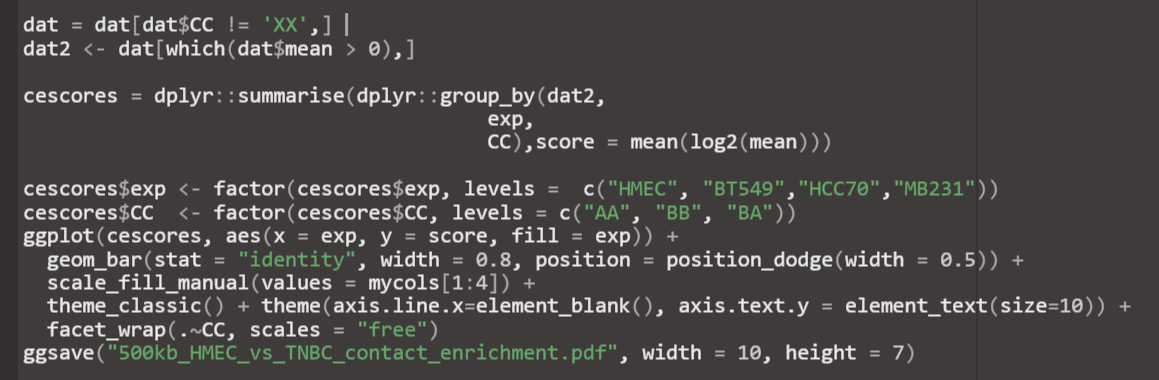
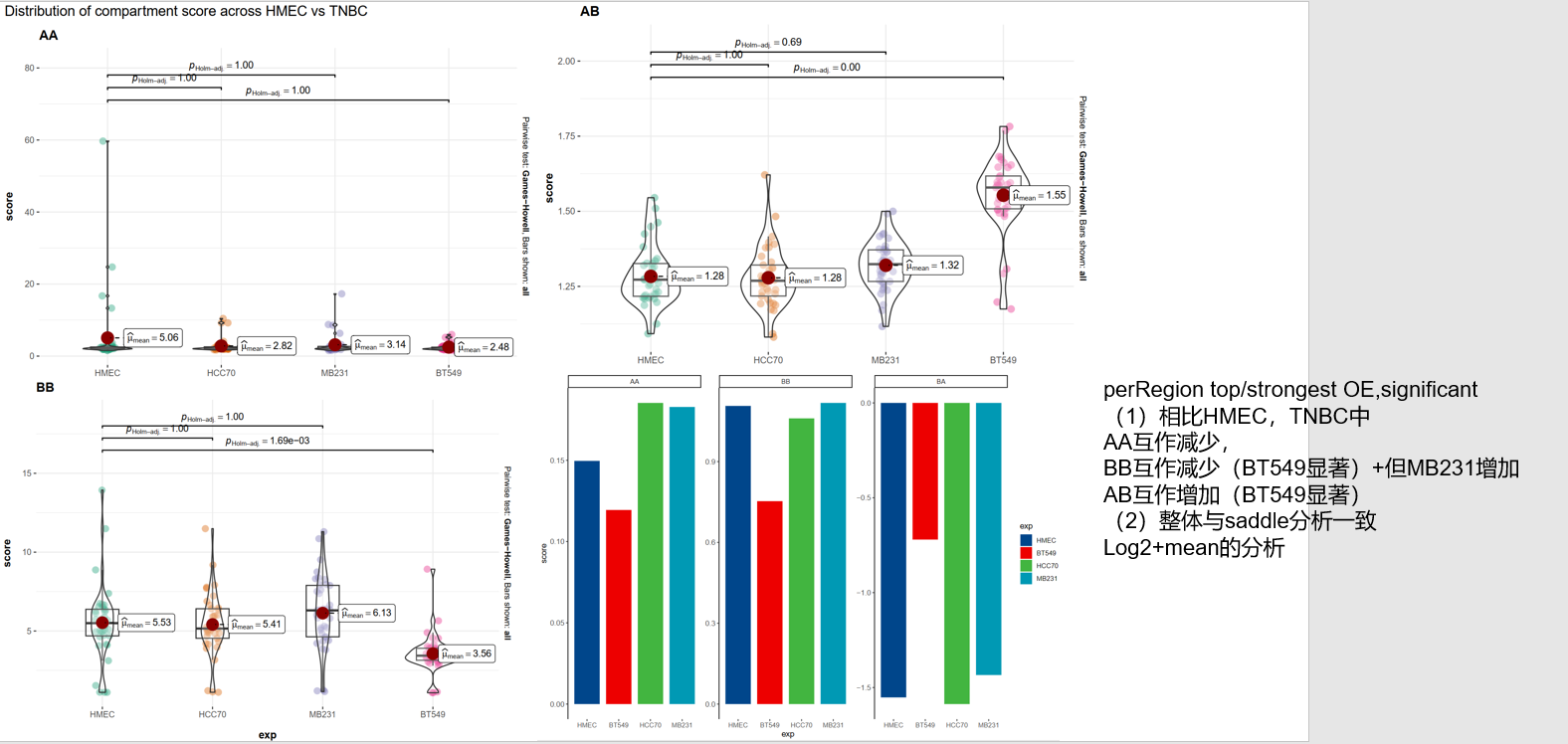
所以右边的条形图实际上就是左边箱琴图的log2,然后右边确实是进行了mean取值将所有的数据,所以理论上可以将左边的数据再进行log2,两次log2;
现在都使用原始的score来计算,基本上数据能够接近以及符合了;

但是实际上分析的时候还是会发现有差异的地方
比如说左边的AA区室中HCC70实际上分数是大于HMEC的,但是在条形图中就会发现MB231是最大的,然后才是HMEC或者是HCC70,
BB区室倒是差不多,然后AB区室分析也是差不多

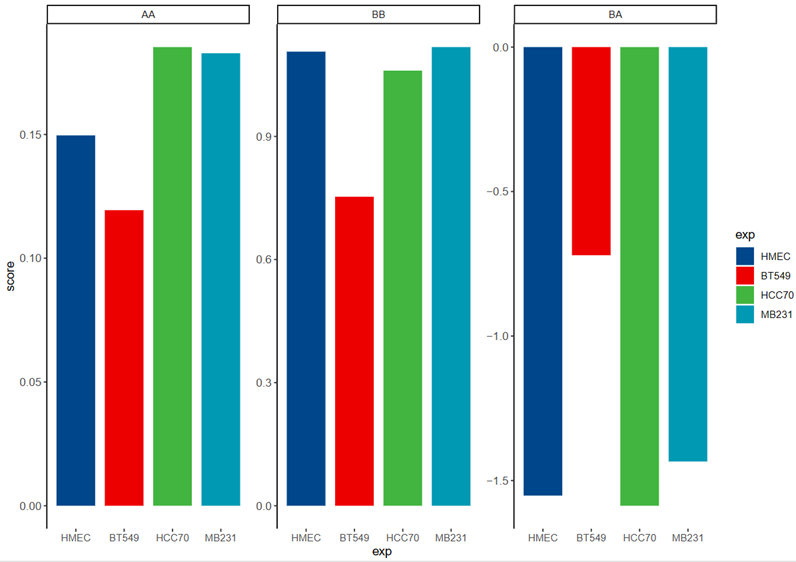
左边是OE score,右边是原来脚本中输出的实际上的log2score
但是因为效果与箱线图不一致,所以还是选用箱线图,
所以我就按照箱线图的数据自己改写了代码,取得还是top的mean,只是为了保持一致,那效果就和箱线图一致了
分析的时候可以说saddle是大致判断,因为本来就分不清楚AB区室,所以用这个箱线图可以更清楚表示,或
②排除异常chr区域,直接排除全部,或者是AA,BB,AB展示中各自排除异常的那一部分
所以可以排除这两条chr之后再绘制score分布图,
当然绘制的还是top20%,那怎么解释,为什么要选取这些区域,
topAB区室区域即strongest,具有显著生物学意义
最终还是用配套数据log,
以及将contact enrichment和这一块合并
絮絮叨叨以上结束
3,AB compartment switch:
(1)使用dchic(https://github.com/ay-lab/dcHiC)分析转换区室情况,
运行dchic主要脚本见run_dchic.sh(https://github.com/MaybeBio/HiC_pipeline/blob/main/pipeline/hic%20%E5%9F%BA%E7%A1%80pipeline%E9%AA%A8%E6%9E%B6/run_dchic.sh),其实就是官方demo中的同名脚本,只要修改个别参数即可;

主要修改dchicf.r的路径(修改成dchic软件安装之后最外层的dchicf.r脚本路径),以及输入文件input.HMEC_TNBC.txt(使用hicpro原生输出数据,本项目使用raw中的matrix+bed),rep分类要分类好;
当然其实这里也有一个问题,上游saddle部分分析区室使用的是mcool文件,虽然说GENOVA也可以使用hic以及hic-pro数据格式;然后这里使用dchic却使用hic-pro数据格式了,虽然这里也可以使用cool以及hic数据格式;就感觉数据跳来跳去,没有一个定数,没有一个是从一而终矫正好的?
如果上游不使用hic-pro,而是使用其他数据格式hic或cool,参考https://github.com/ay-lab/dcHiC/wiki/Pre-Processing-Data进行转换,并且参考issue等取经。
另外注意input的txt中matrix+bed最好移除异常chr数据(这在上游hicpro的处理中就可以进行,只保留1-22+XY),另外也建议移除chrY,依据官方的指示:


(2)dchic输出结果分析:
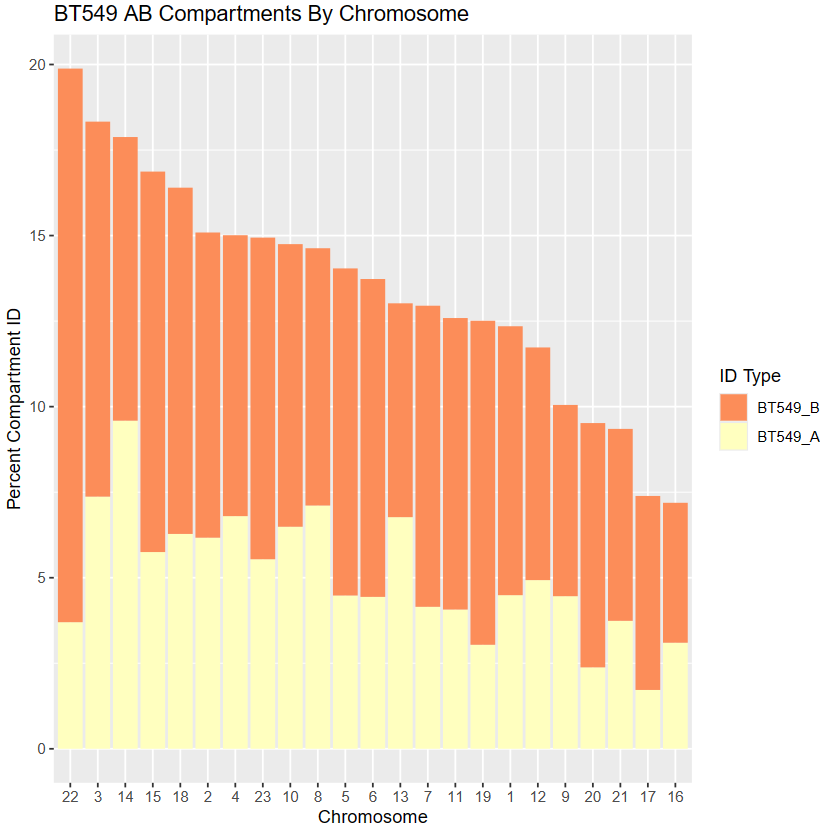
①作为输出,dcHiC创建了两种类型的目录。第一个是原始的PCA结果,在以输入文件的第三列命名的目录中。其中一个是为每个输入Hi-C配置文件创建的;在内部,将有“intra_pca”或“inter_pca”目录,这取决于用户是否基于染色体内或染色体间的相互作用和每个染色体内部每条染色体的原始PC值来指定分区计算。
如下图为例_pca为结尾的文件夹
②第二个总括目录称为differalresult,它包含用于差异结果的目录(在任意数量的参数设置上)。这些目录名在-analyze pcatype选项(执行差异调用)dcHiC下指定,用户在其中表示要进行分析的——diffdir。全局的differalresult目录下可以存储多个目录,具有不同的分析参数;
同样,如上图所示的第一个diff文件夹;
在每个diffdir中,有原始的分区结果(“expXX_data”)和两个PC输出目录PcOri和PcQnm,以及组合和分位数标准化的分区结果。最后,将有一个目录fdr_result,其中包含不同的分区结果、loop结果和子分区结果。在fdr_result中,sample_combined文件包含所有XX细胞系跨重复的平均PC值的完整bedGraphs,以及表示该隔间bin的Hi-C实验之间变化的显著性的最终调整p值。sample_combined filter的文件包含相同的信息,通过p值截止进行过滤。

如上图所示,最下面的4个exp对象就是_data结尾的文件夹,有原始的分区结果
然后最主要的文件夹就是fdr_result,其中的sample_combined就在其中;
按照官方文档说明,filtered文件含有的是p值过滤之后的文件。
其他的文件输出就是:
gene富集文件夹中:anchor也许可以用,要清楚这个是怎么产生的
viz文件夹中:

vizIGV_intra中有IGV的html文件
有这个的可视化IGV,
可以作为全局可视化比对区室转换,也可以搭配其他track(除了其他组学的track,在dchic的其他结果产出部分也有bedgraph之类的文件可以试一试);
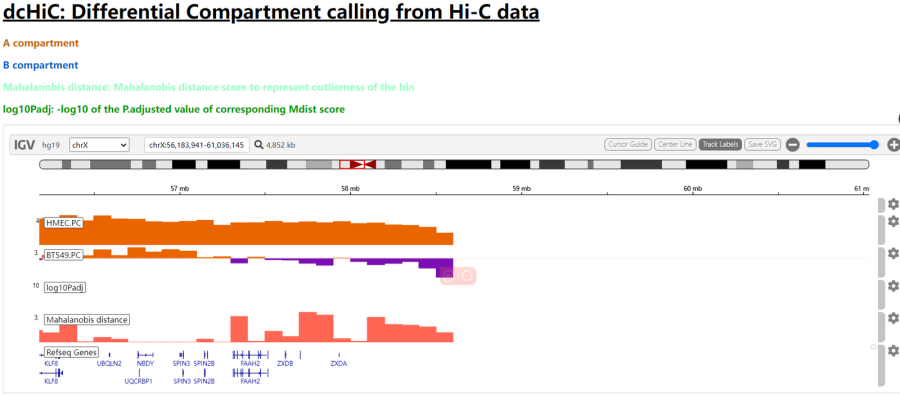
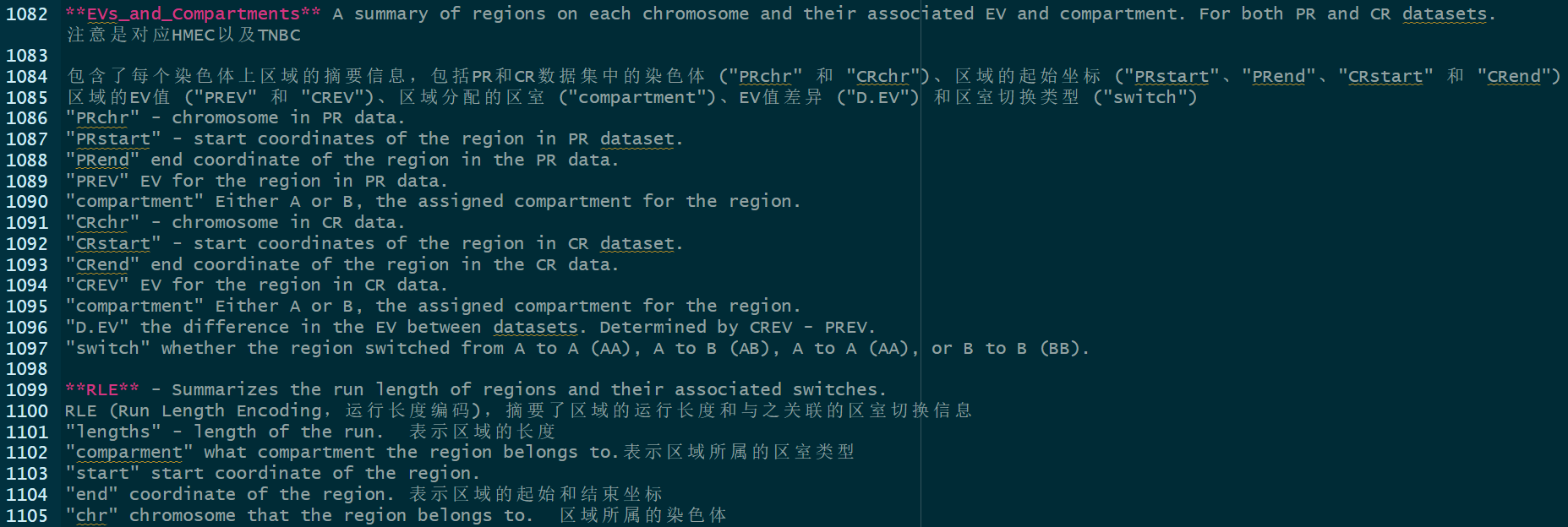
至于用于区室转换分析的输出结果文件,只要记住
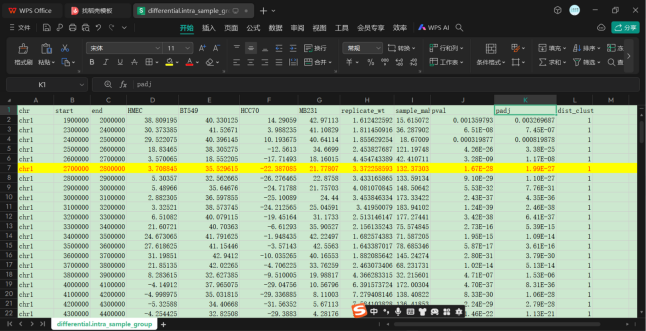
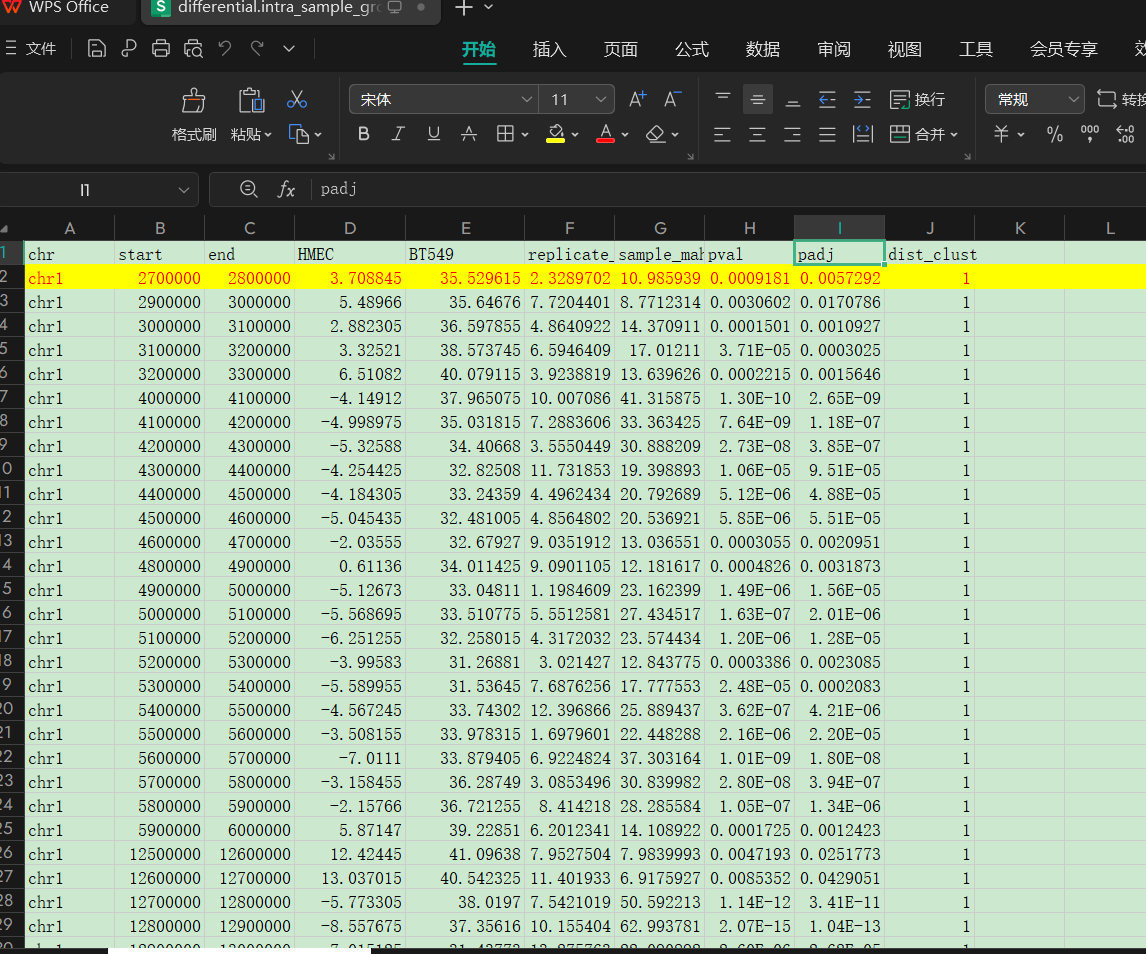
**DifferentialResult/HMEC_BT549_100Kb/fdr_result/differential.intra_sample_group.Filtered.pcOri.bedGraph为经过dchic默认padj过滤的结果文件(fdr阈值)——filtered,
DifferentialResult/HMEC_BT549_100Kb/fdr_result/differential.intra_sample_group.pcOri.bedGraph没有经过过滤,可以自行设置阈值——full**;
我们一般选用full原始文件,然后自己筛选阈值处理;
(3)dchic过程分析模块:转换的AB区室
**注意以下分享的代码图表都是02_Analysis_dchic.Rmd(1vs1,即HMEC vs BT549)脚本中的图表+对应row数,但是实际操作时候使用的是02_Analysis_dchic_1vs3.Rmd(1vs3,即HMEC vs TNBC)脚本,但是没有放上对应的图表以及row数,注意要参考02_Analysis_dchic.Rmd的注释来执行并理解02_Analysis_dchic_1vs3.Rmd,对应部分逐行参考注释并理解;
依据线索:原始脚本PDXHiC_supplemental/Fig5_HiC_ABcompartments/01_Analysis_dcHiC.Rmd——》拟合注释1vs1的02_Analysis_dchic.Rmd(HiC_pipeline/pipeline/hic 基础pipeline骨架/02_Analysis_dcHiC.Rmd)——》改进之后1vs3的02_Analysis_dchic_1vs3.Rmd(HiC_pipeline/pipeline/hic 基础pipeline骨架/02_Analysis_dcHiC_1vs3.Rmd)**
拟合原脚本参考https://github.com/MaybeBio/TNBC-project/blob/main/2%2CCompartment/01_Analysis_dcHiC.Rmd,后来拆分为02_Analysis_dchic.Rmd+02_Analysis_dchic_1vs3.Rmd
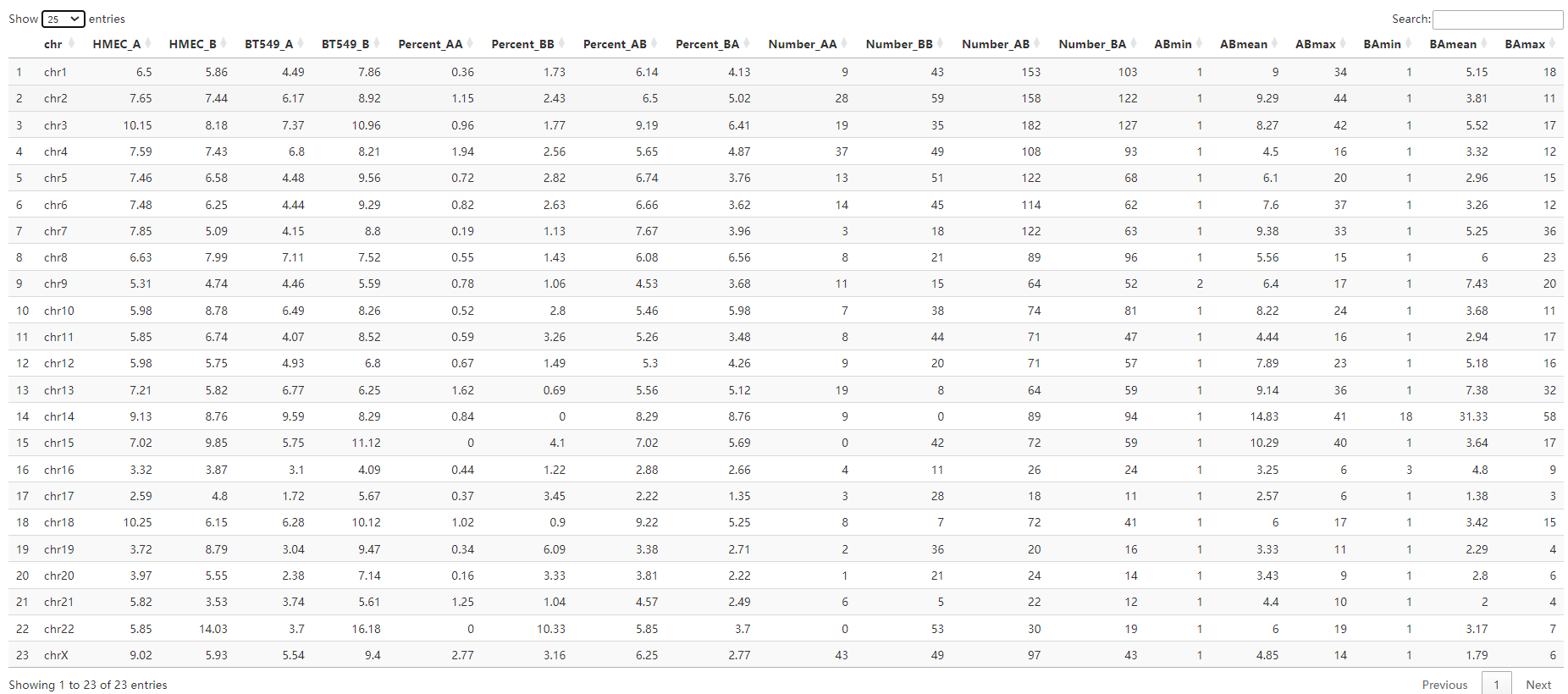
①各种chr展示表格:

打印出了proportioNumber_AB_chromosome:每条染色体上AB区域变化情况
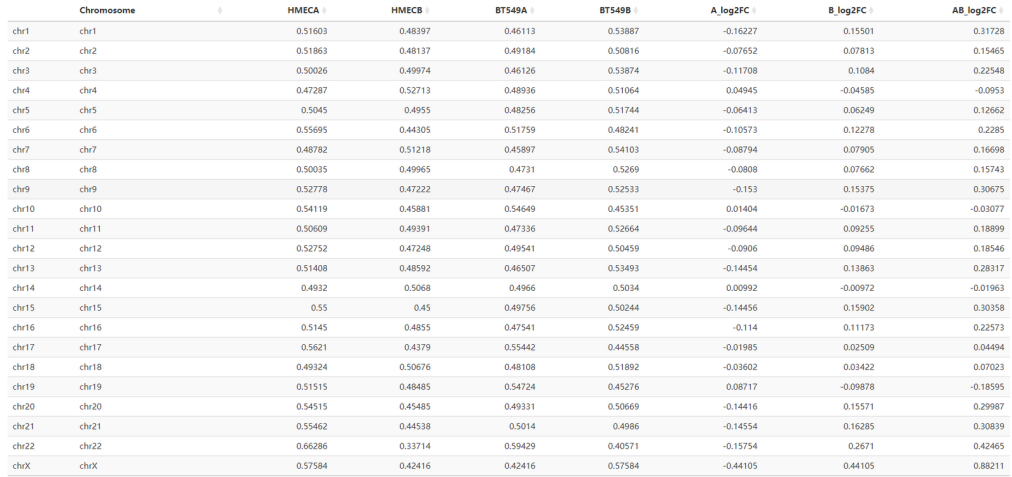
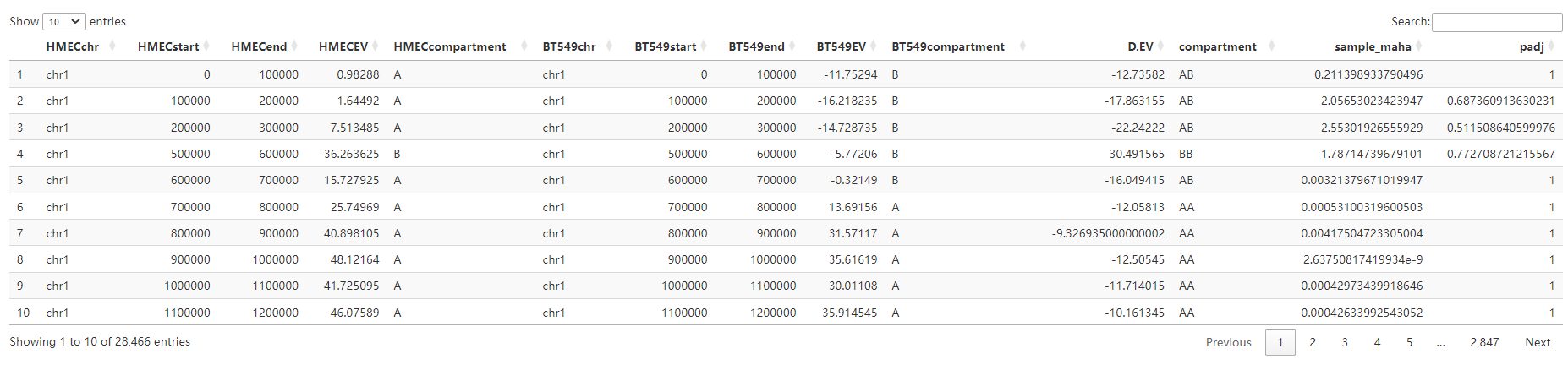
对这张表格的解释分析:使用的数据是full,即未经过padj0.1过滤的所有原始区室EV值数据,但是去除了问题区域
“HMECA”, “HMECB”, “BT549A”, “BT549B”:对应的各自条件下的AB区室比例(注意因为使用的是原始的全局数据,并没有经过padj过滤,所以比例上合起来是100%的)
“A_log2FC”, “B_log2FC” :A或者是B区室各自在HMEC与BT549中的log2 FC比例变化
“AB_log2FC” :A/B区室比例在HMEC与BT549中的差异
AB_log2FC = log2(HMECA / HMECB) - log2(BT549A / BT549B)
**当然这里也有一个问题,就是到底应该使用全部的区室数据还是使用padj过滤之后的区室数据作为展示以及后续的分析?**其实都可以,按照自己数据分析需求;

还打印了AB_conditions_assigned,即区室变化的padj过滤之后的原始score数据文件,主要有D.EV指;
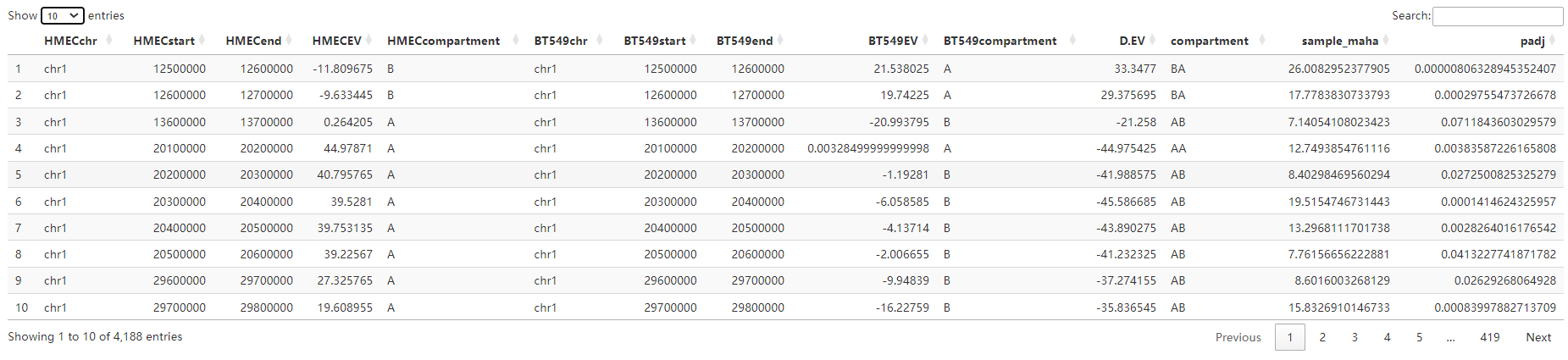

以及AB_conditions_assigned_all,同上是未经过padj过滤的数据

以及一张AB_summary(已经分析出区室转换AB比例的chr表)
**使用dchic时一直有一个问题没有去深究:转换的区室如何识别整合,如何对应?
一方面是dchic识别整合的问题,另外一方面是处理脚本如何对应的问题——我感觉RLE编码那一部分是有关的。**
有些issue可窥一斑:
有些关于padj以及显著的问题需要解决,
https://github.com/ay-lab/dcHiC/issues/53
主要是dchic中的padj的问题,
首先区室信息即是A或B肯定是能够直接从原始数据的sign符号上获得的,这个不需要靠p值,但是问题是我们关注的问题从来不是单一样本中的AB区室,而是区室转换即AB区室转换,而区室转换实际上就有显著与否这个指标了,所以padj应该是用在衡量区室转换是否显著上;
至于如何判断区室转换是否显著(即如何处理单个res下的bin,到AB区室,那是dchic内部算法问题,可以参考上面那个issue),
总之比较的区域是一致大小的,是相同大小的bin区域
最重要的是dchic本身就是用来识别分析显著区室转换的
②区室转换的核型图KaryoploteR Plots:

简单来说就是将每一条chr上的AB区室以及score值映射到基因组chr坐标上的图
KaryoploteR Plots and Per Chromosome AB Compartments and EVs
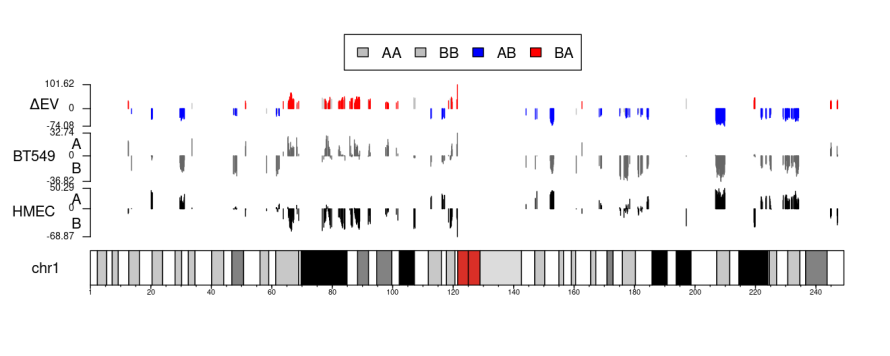
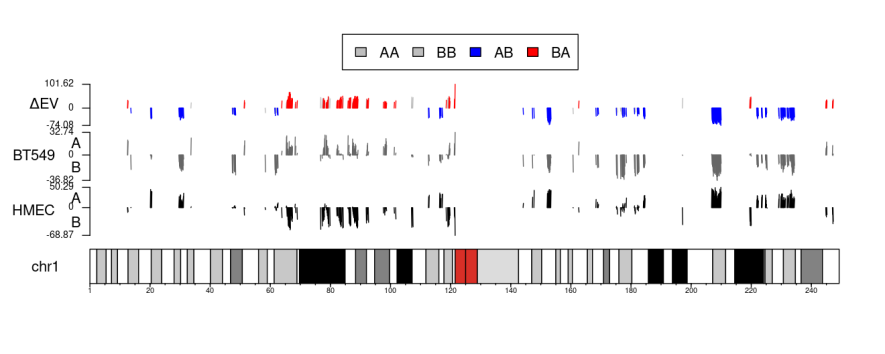
前者是x0start+x1start,后者是x0start+x1end,从效果上看两者没有区别,图都保存了
需要搞清楚这个图的意义以及解释,主要就是下面的chr bar,
至于为什么区室有些是空的:
一方面是测序的问题有些区域没有测到(这个可以在最前面的juicebox热图中观察到),然后还有就是排除了一些区域(端粒以及着丝粒区域等等,当然要看一下下面的ref里面也有没有排除对应的问题chr区域)
最后就是使用padj过滤的问题,所以比较显著的区域如上:

除了全部数据的,还有仅含有EV值的
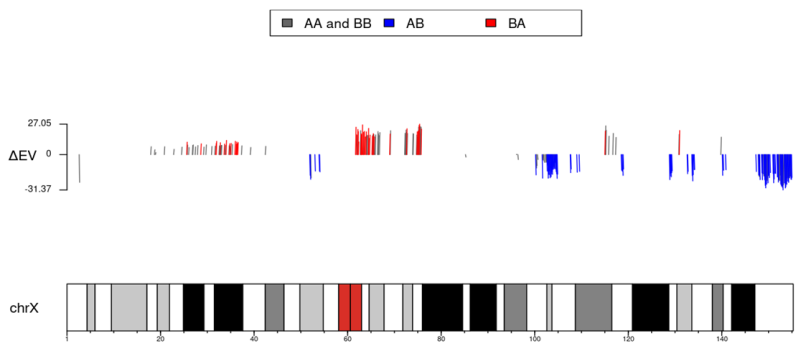
③区室转换的一些chr统计图:
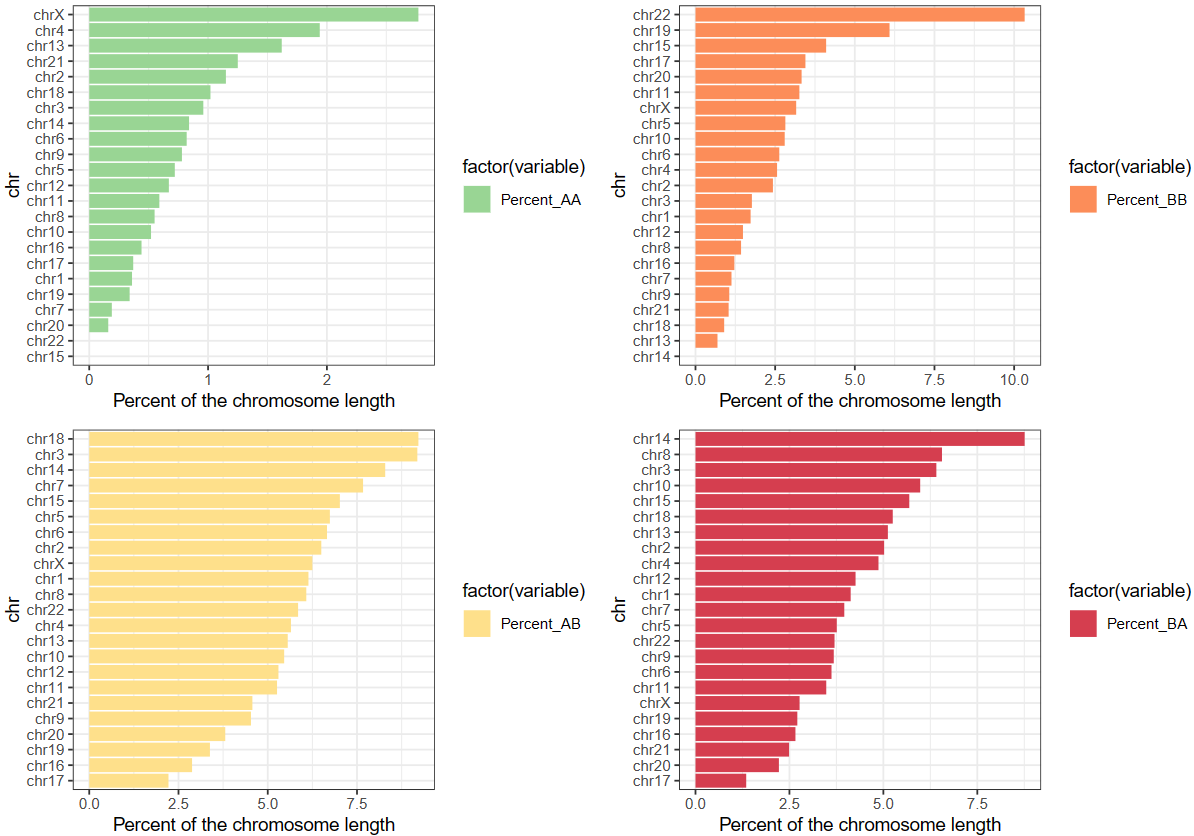
bar条形图

HMEC vs TNBC各自chr上AB区室的比例——展示静态
HMEC vs TNBC对应chr区室AB变化的比例——动态变化
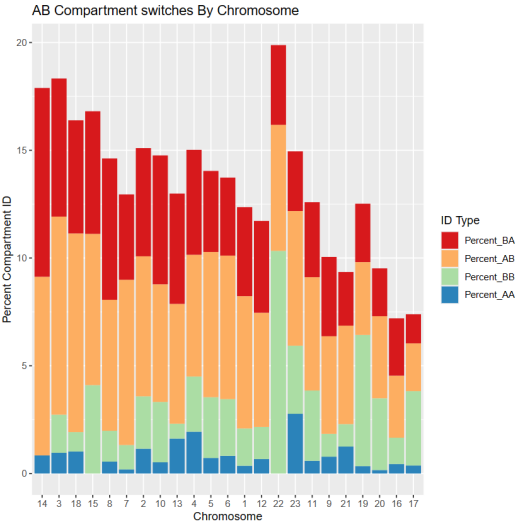
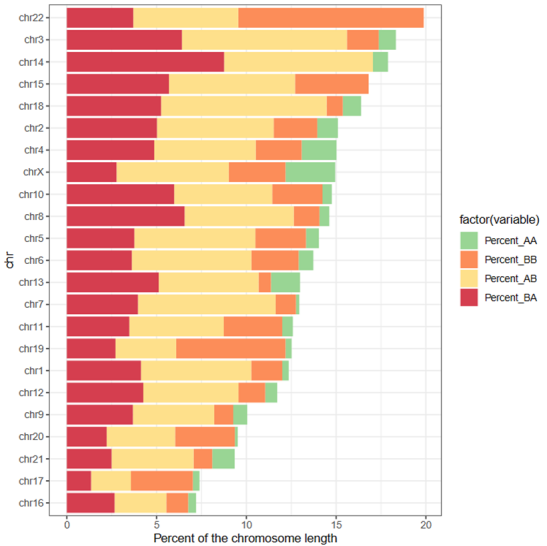
绘制了proportion of a selected AB compartment change
就是AA,AB,BA,BB等转换区域分别对应各自chr长度的比例,
pie饼图:


可以使用CNSknowall等相关在线网站进行绘制
桑基图,也就是所谓的流图flow chart:
这部分需要自己编写脚本,利用AB_summary中的数据构造了绘制桑基图冲击图所需要的数据框,主要是显著的区室转换部分的Number的数据
以在线网站rawgraphs2.0为例
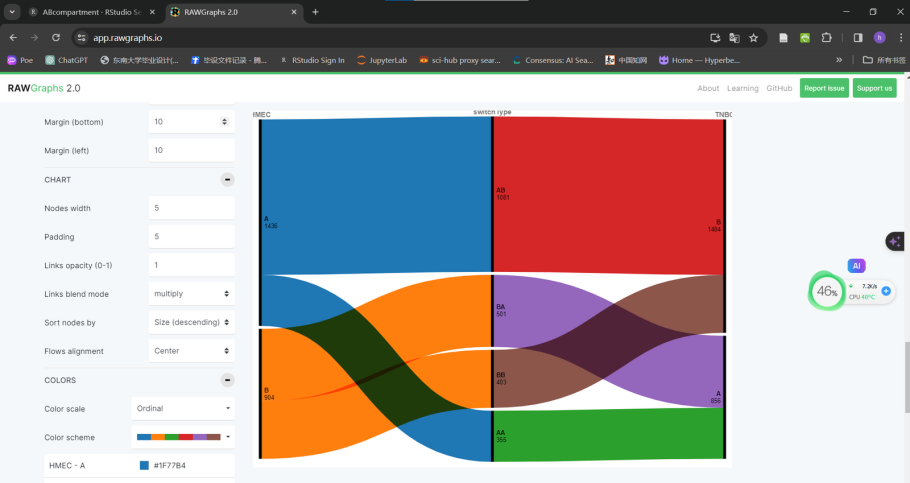



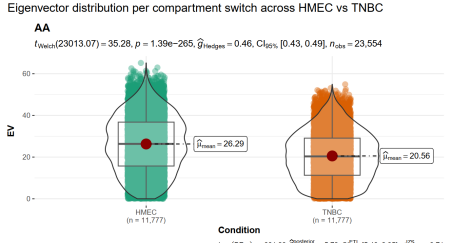
④EV值(特征向量值,本质上就是区室score之类的)在各种区室中的分布:
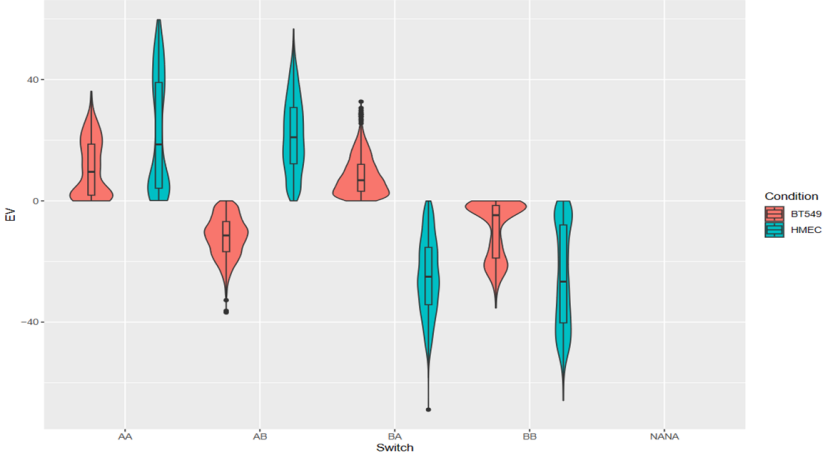
可以从中分析区室变换的趋势,可以使用
(4)转换的AB区室与相关gene分析:
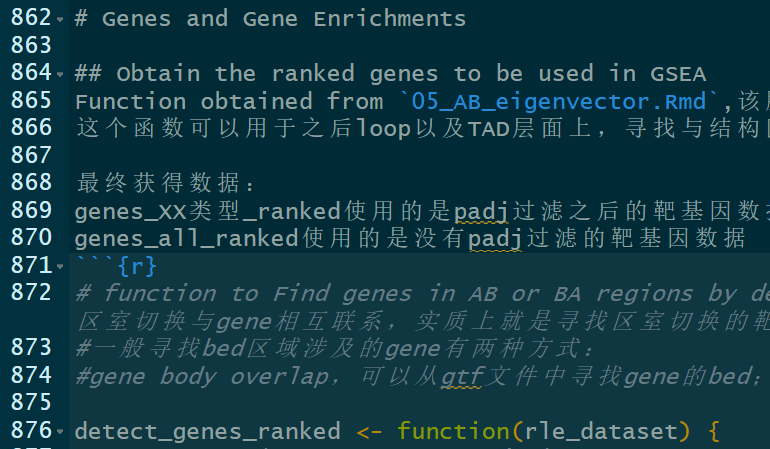
①重点在于找到与区室转换区域所overlap的gene:
有2种方式:
1种是直接gene body(可以从gtf文件中获取器bed文件,基本上只要gene,start,end)与区室转换区域bed overlap即可,
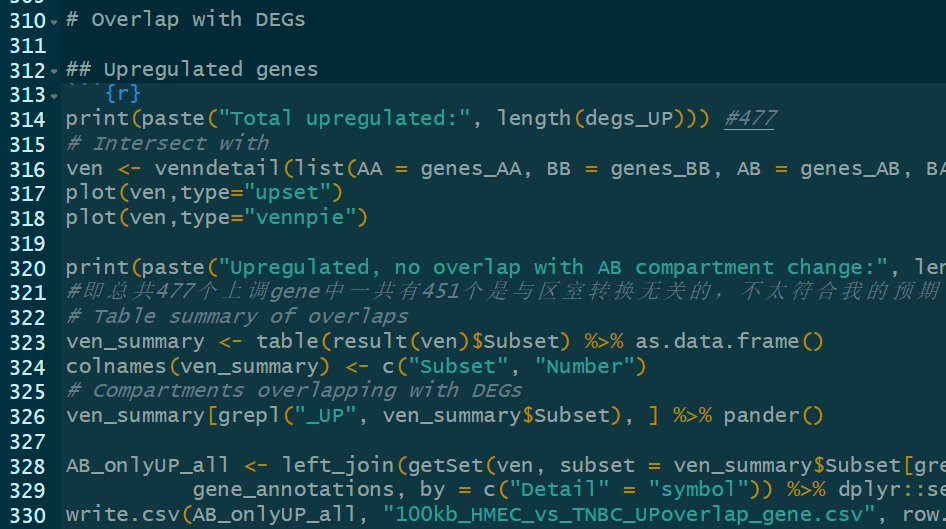
第2种是定义启动子promoter(即获取gene的TSS位点,一般依据分析经验是定义其上下游1kb左右),然后启动子bed与区室bed重叠;
至于重叠,其实有很多工具可以使用,比如说linux工具bedtools,或者是一些常见R包的开发函数,比如说是chipseeker或者是chippeakanno的一些peak/bed处理函数;
参考脚本02_Analysis_dchic.Rmd中定义的函数,后续还可以再用于loop以及TAD区域涉及的gene(因为本质上都是bed文件之间的处理):
其中最麻烦的问题在于gene id的转换,可以参考我的另外一篇博客https://blog.csdn.net/weixin_62528784/article/details/142170917?spm=1001.2014.3001.5501,
首选ensembl+bioMart,其次是clusterprofiler的bitr函数;
最后获取的gene信息:
相应的gene表格以及对应参考基因组信息bed(下图是1vs3.Rmd中的,并非1vs1,见该子小节处置顶)
获取的data:
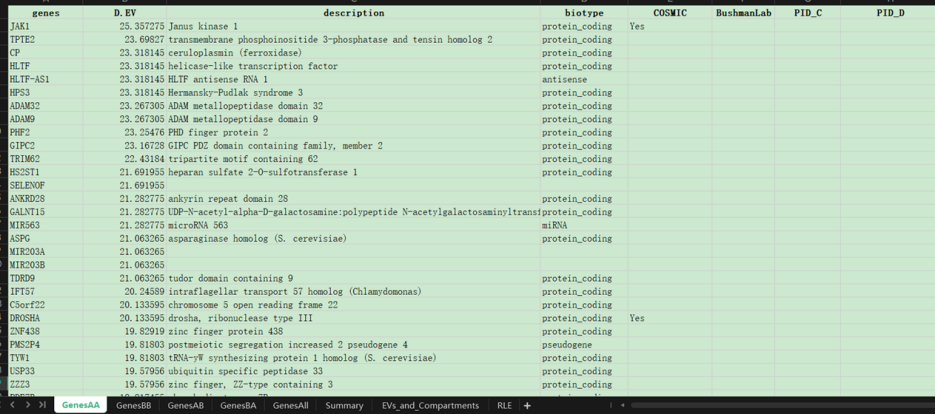

重点在于以上这几个底部的sheet集,是后续gene分析的重点
获取的bed:

②对这些gene做表达分析,并且gene表达与区室转换的指标联系起来:
因为获取了gene表达数据,所以可以做常规的RNA-seq分析,此处请参考我的另外一篇博客:
https://blog.csdn.net/weixin_62528784/article/details/142218107?spm=1001.2014.3001.5502
此处参考脚本
指标转换比如说:
看看4种区室转换类型(AA,AB,BB,BA)对应的区域,这些区域转换过程中EV指的变化趋势,即D.EV,比如高EV是强A,低EV是强B,看看转换类型是强弱AB与强弱AB之间怎么样的转换?
看看4种区室转换类型(AA,AB,BB,BA)对应的区域所overlap到的gene,即所涉及的gene,其表达变化趋势,比如AB是表达下降,BA是上升等,可以绘制1个聚类热图;行是gene,列是样本?
看看4种区室转换类型(AA,AB,BB,BA)对应的区域所overlap到的gene,即所涉及的gene,其表观修饰信号的变化趋势,比如所ATAC指标、以及H3K27ac等组蛋白修饰信号指标变化如何等;同样可以绘制热图?
看看4种区室转换类型(AA,AB,BB,BA)对应的区域所overlap到的gene,即所涉及的gene,其在比对前后(比如说是TNBC vs HMEC) logFC等指标与这个区域本身指标D.EV之间的关系,可以拟合回归?
实际执行过程中:
做overlap即韦恩图的:

然后可以细致查看具体交集中是哪些gene,比如说有些在AB转换区域又在BA转换区域,搞清楚原理,是不是error?

与DEG做交集分析,只是探索性,也是画upset韦恩图等,实际上就是看看up以及down中的gene具体和4类区室转换区域所overlap的gene有什么关系;探索性而已,不纳入正式分析,前面同理也要思考
相关性分析:其实就是前面,可以纳入正式分析

做富集分析:

画表达热图:实际上是

这里涉及到一个问题,也就是gene id转换的问题,建议上下游统一注释参考等,建议使用ensembl+bioMart。
参考我的另外一篇博客:https://blog.csdn.net/weixin_62528784/article/details/142170917?spm=1001.2014.3001.5502,
对照原始脚本PDXHiC_supplemental/Fig5_HiC_ABcompartments/02_Analysis_dcHiC_genes.Rmd,拟合之后为https://github.com/MaybeBio/TNBC-project/blob/main/2%2CCompartment/02_Analysis_dcHiC_genes.Rmd,调整修改为HiC_pipeline/pipeline/hic 基础pipeline骨架/03_Analysis_dcHiC_genes.Rmd,
直接对照原始脚本+现在处理脚本即可,对照参考注释理解,具体执行还是使用原始脚本,该系列hic系列脚本都这样!
③对这些gene做富集分析
富集分析方面,可以参考我的另外一篇博客;
暂时没写,待更新。。。下面的可以跳过了
下面是dchic的絮絮叨叨处理过程经历以及零碎经验,可以直接跳过——————————————————这是一条分割线
利用dchic进行后续区室转换分析:

首先是安装dchic工具,在dchic环境下
主要教程就在于:
https://github.com/ay-lab/dcHiC/wiki+主页的demos+https://github.com/ay-lab/dcHiC/tree/master/demo+https://github.com/ay-lab/dcHiC/blob/master/demo/README.md,
先看demo
在git之后在/mnt/disk4/haitao/software/dcHiC/demo/
再看/mnt/disk4/haitao/software/dcHiC/docs/
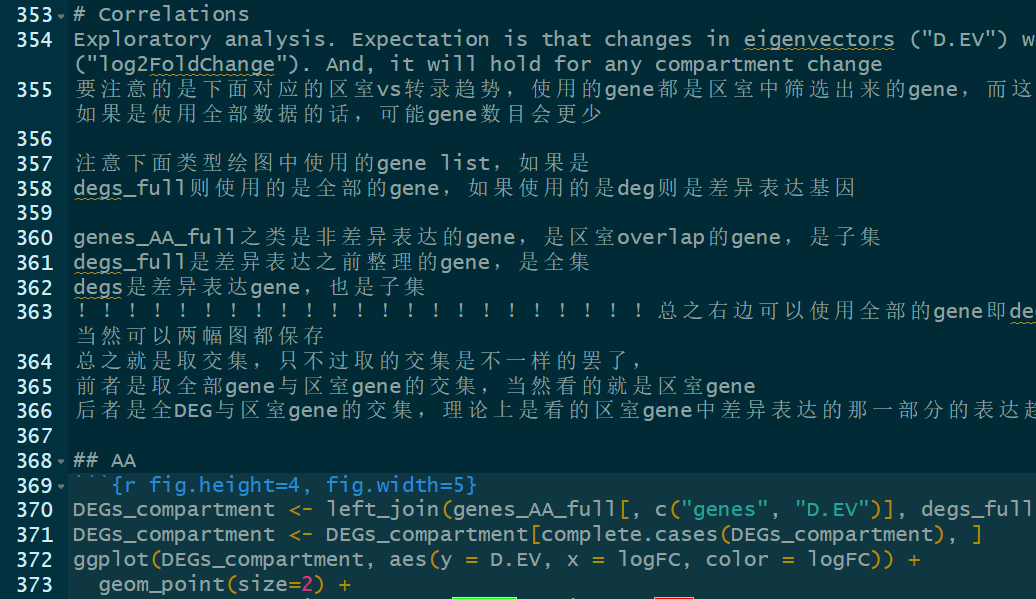
docs——》demo——》wiki,
demo是用来参考的,但是运行脚本以及修改参数的时候注意要在最外层,不要在demo中,
在执行的时候:https://github.com/ay-lab/dcHiC/issues/50
应该是使用线程数的问题:只使用c、pthread,然后分别修改成1;
然后遇到了异常chr的问题:
https://github.com/ay-lab/dcHiC/issues/69
https://github.com/ay-lab/dcHiC/issues/71
试试看移除chrY之后能不能解决:

应该就是chrY的问题,但是为了防止文件出错失误无法挽回,
建议是对于bed文件copy一个副本,在copy上删除chrY,然后之后再在input的文件里使用副本文件;
但是坐标index会出现在matrix文件上,所以又建议matrix也copy一份,然后删除index,最后都一起copy在input文件中;
cp HMEC_rep1_100000_abs.bed HMEC_rep1_100000_abs_copy.bed
awk '{if($1!="chrY") print $0; else print $0 >> "chrY_removed.bed"}' HMEC_rep1_100000_abs_copy.bed > HMEC_rep1_100000_abs_copy_no_chrY.bed
先不修改matrix文件,主要是确实如果是上三角矩阵的问题的话会有另外麻烦;
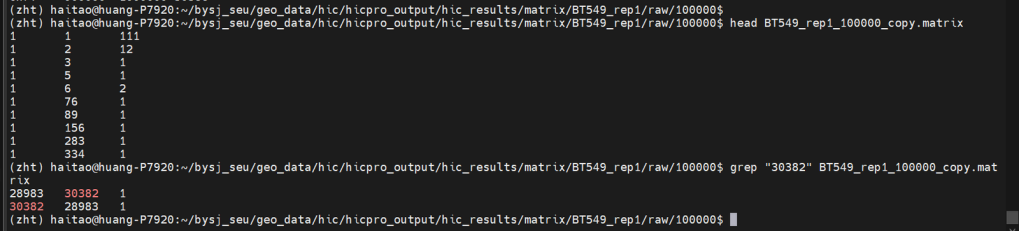
https://github.com/ay-lab/dcHiC/issues/88,
报错了,我觉得还是先解决上三角矩阵的问题:
其实一个对称矩阵转换成上三角矩阵很简单,打印出$1<=$2的行即可
awk '$1 <= $2 {print $0}' $matrix_file > $new_matrix_file
先解决上三角矩阵,再解决chrY等问题
现在是解决了上三角矩阵的问题+原来的bed文件(未去除chrY等),再运行试试看
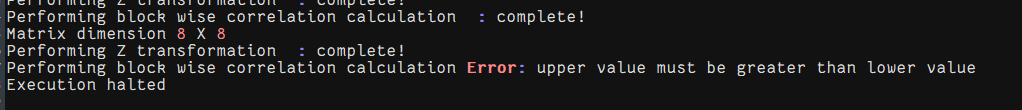
还是同样的报错,解决上三角matrix(未清理chrY的index)+清除chrY的bed文件

还是同样的报错
接下来对matrix中剩余的index进行处理,就是
对照chrY_removed.bed的第4列,只要是upper_matrix第一或者第二列有出现在前面chrY第4列的直接删除,保留下来的matrix文件
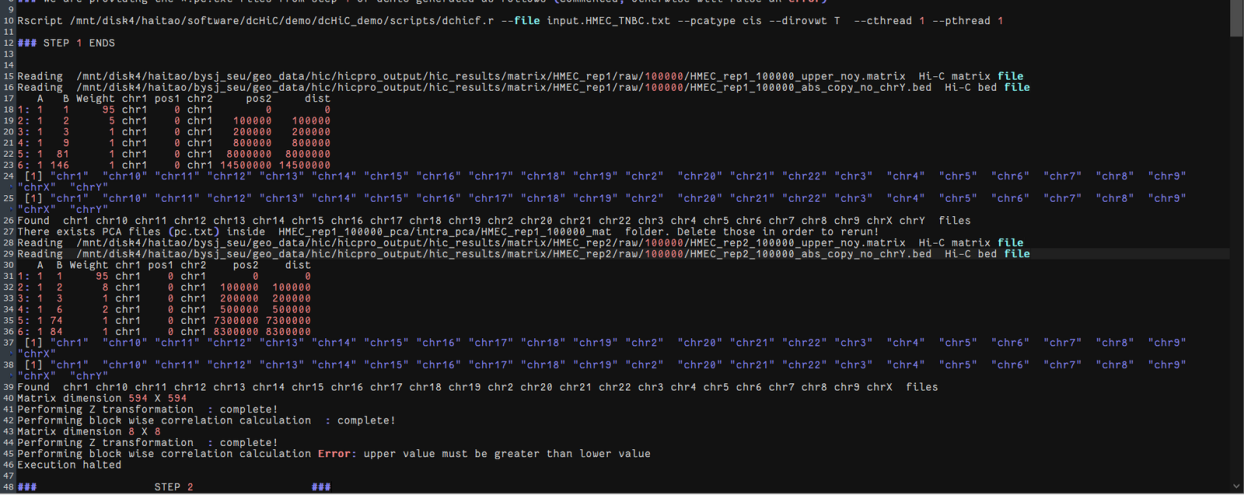
还是同样的报错,而且看前面的信息chrY还是有的
bed文件用cut -f 1 xxx.bed | uniq检查过是没有问题的了
注意要清理前面跑的几次的文件夹,里面的文件可能残留chrY等信息,会被新线程直接使用,这样会报错;
当然,对于hic-pro还有上游配置文件config文件中使用upper还是complete的问题,之前操作时是complete,所以这里我处理hic-pro上游文件才要这么麻烦地去处理上游matrix或bed文件,我在想,一开始就不改默认设置upper是不是就很轻松,大概后面跑第2次就改过来了。
首先所有的脚本都是对照dchic官方所提供的demo中的脚本run_dchic.sh
运行的基础是R脚本dchicf.r,也是官方所提供的;
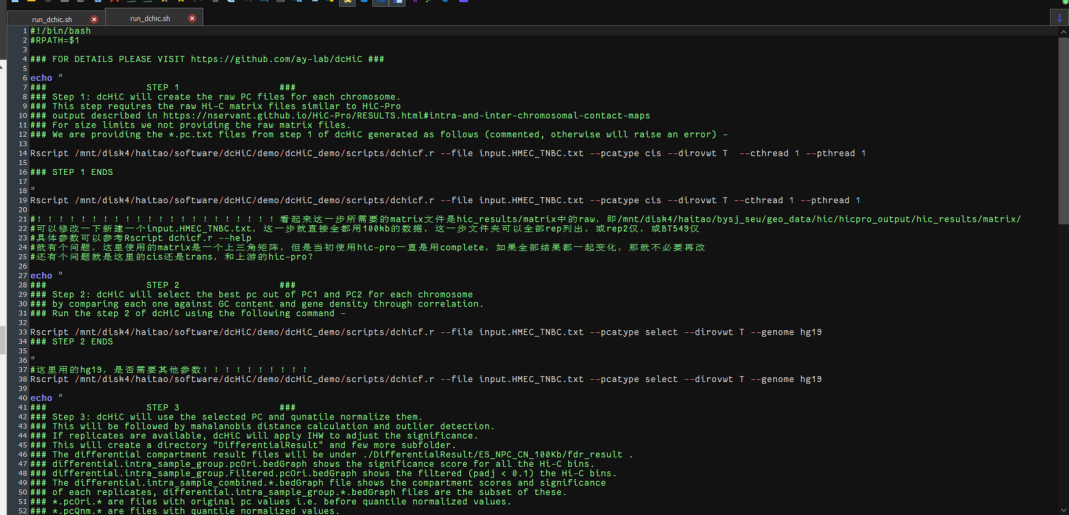
大体上我只是在input.txt文件上进行了修改,
按照HMEC vs BT549/HCC70/MB231的3对1vs1的比对模式分别创建了3个文件夹,分别运行得到1vs1的比对输出结果;
然后就是每一个比对中都只执行了部分步骤(关于loop分析相关的step4,5都没有执行,至少在这3个文件夹中)
操作过程中遇到的一个比较纠结的问题:
intra的bedGraph与fdr中的filter的bedGraph文件哪个作为下游dchic分析脚本的输入?
因为数据量的问题,以及使用intra的文件过程中一直有一个padj的阈值设置的模糊问题(但是这个intra文件实际上又并没有使用上dchic初始call compartment时候设置的阈值的过滤,所以如果使用filter文件,也许就可以跳过这一步,直接使用全部的bedGraph——主要就是看不懂原来分析脚本中对于padj的设置以及处理;
但是intra那个文件,实际上又是和上面那个html的IGV是在同一个文件夹中的;
其实就是文件设置问题:
fdr中的filter文件,原始bedGraph文件
intra的bedGraph文件
filter与原始的bedGraph文件是p值过滤的结果,用的就是dchic初始脚本中
另外注意到实际上原始的bedGraph文件与intra中的文件行数是一致的;
https://github.com/ay-lab/dcHiC/issues/53
另外文件的问题:

ori文件是能够用分数表征AB的,但是qnm文件就是定量化之后的
还是回到了Padj设置的问题上
所以首先分析下游中使用是不是直接AB值,如果是,就不用Qnm数据,使用ori数据
那就是:
fdr中的filter文件,原始bedGraph文件
intra的bedGraph文件之间的问题
注意到fdr文件夹中还有group以及combined的数据



可以看出group和combined的数据基本上是一致的,只不过combined中将group内部的rep数据也展示了出来;
而在具体下游分析时不用考虑rep内部数据
所以使用group数据来替代combined数据
那就全看group数据
因为Qnm的数据前面比对已经发现和intra是不一致的,而且也不推荐使用
就比较group-ori数据和intra的数据
首先分析group-ori-没有filtered的数据和intra的数据:

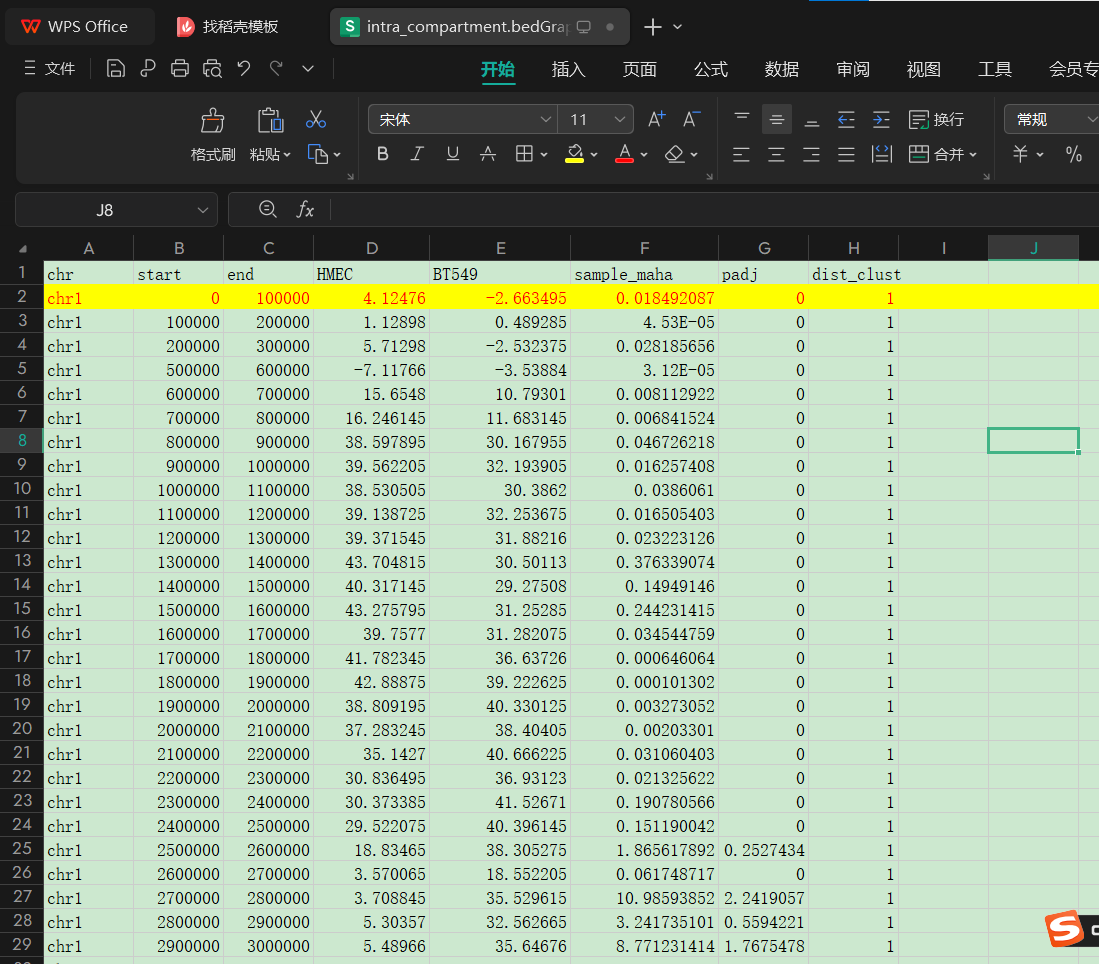
可以看出来inta的数据和group-ori-没有filtered的数据基本上是一致的
后者有p值,两者之间的padj略有差距
那么实际上下游分析脚本使用的输入文件
两者都可以:inta的数据和group-ori-没有filtered的数据
但是dchic下游分析的脚本上就涉及到了阈值filter的问题,
理论上来说如果上面两个数据一致的话,group-ori-filtered的数据实际上就是intra的filtered,是可以直接使用从而跳过下游分析中阈值的这么一个问题
但是不清楚filtered的标准是什么,所以相当于是一个黑箱
就是因为是黑箱,所以下游分析的时候R脚本中才自己设置了一个阈值,而且用的是padj
如果能搞清楚原始dchic本身输出中filtered的标准,评估可行性,实际上就能够直接使用这个文件了;

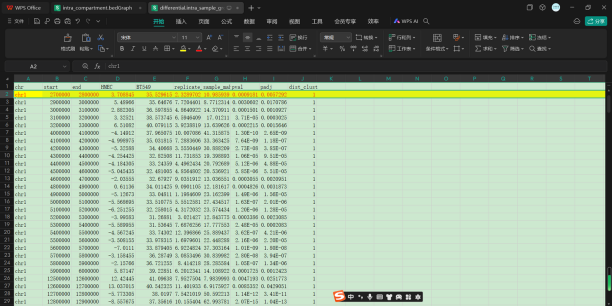
还是一致的,在group-ori-filtered&intra中
https://github.com/ay-lab/dcHiC/issues/74

另外一个问题就是1vs3中的数据与1vs1中的数据对应问题
下面是group-ori-filtered中HMEC vs TNBC与HMEC vs BT549的比对
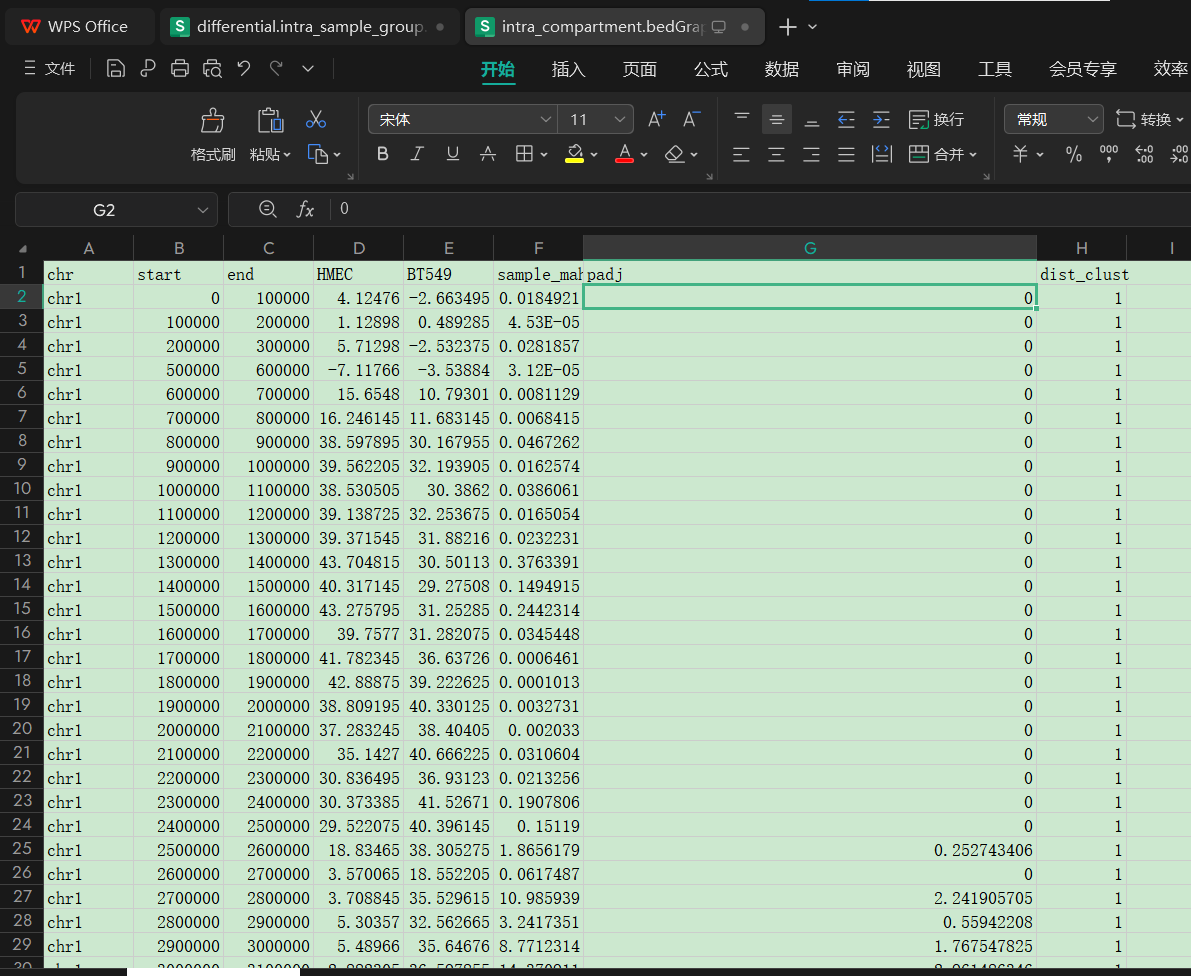
可以发现rep的值是一致的,但是p值无论是adj还是val都不一样
所以不能使用这个1vs3的分析,不然p值都公用也许检验的标准不一样
所以做3次,
使用1vs1的group-ori-filtered(阈值未知),或者intra自己下游设置阈值

https://github.com/ay-lab/dcHiC/issues/97
查看源码dchic.R或者是demo中run_dchic.sh脚本

这里说的是padj<0.1,如果没有记错的话下游分析脚本中使用的padj是0.3,也就是说实际上官方所筛选出来的dchic数据实际上更严;
那还是使用自己的脚本分析,然后在阈值分析上筛选出<0.1的
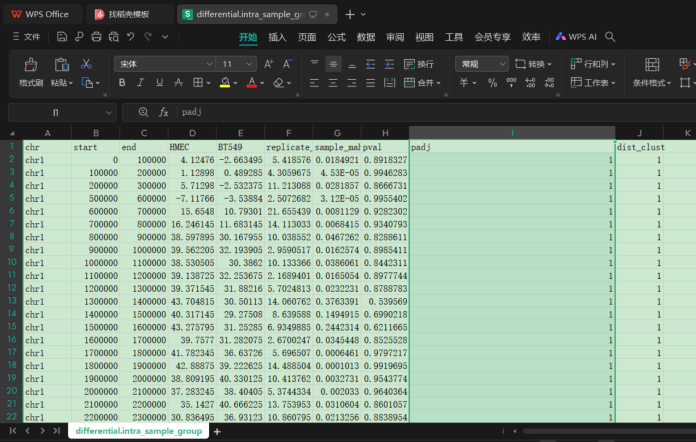
左边是intra,右边是未过滤
所以如果是自己设置阈值过滤的话,其实使用intra和未过滤文件就有一个问题,就是:
明明是理应同样的一份文件,为什么计算出来的padj上却完全不一样
其他文件也不一一样,所以很麻烦,其实计算的时候会出问题
要么就是使用未过滤文件+自己设置cutoff,要么就是默认使用0.1的过滤文件
intra还是慎用!
查看源码分析为什么会有差异,

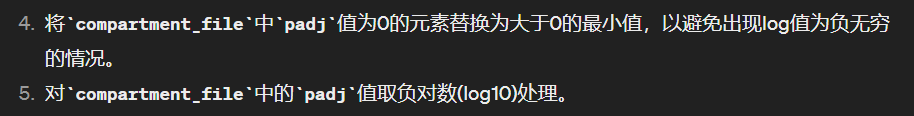
所以实际上intra的值是原来未过滤文件的一个lg化版本+处理特殊0值
查看查看各个脚本之间的区别以及差异:
demo中的脚本需要仔细对照,实际上并没有太大区别:


再看脚本输入选项:
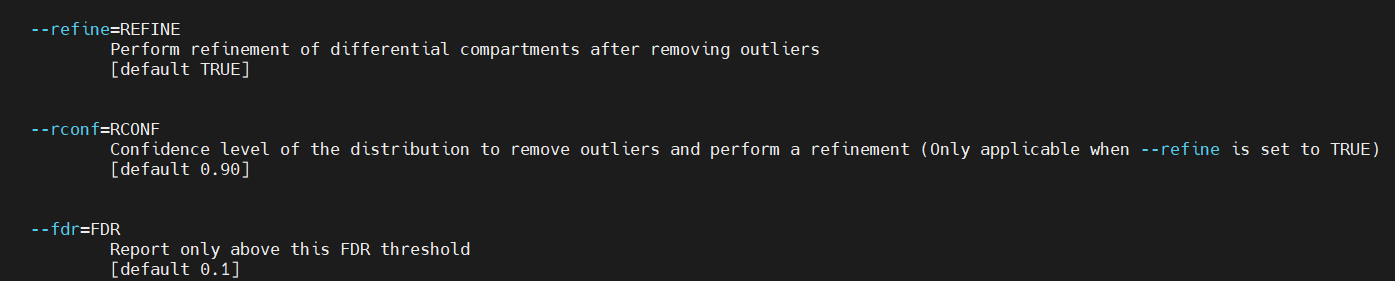
脚本里面能够输入阈值筛选,而且是默认设置的0.1
所以实际上我们脚本之间没有区别,因为fdr等阈值都是default的,不放心就实际加上去
综上问题来了:
本来过滤筛选的是padj<0.1的显著,这个参数是默认的(我们可以改,在未过滤的文件中,使用其他的参数,但是是针对padj)
然后对于intra文件,实际上padj已经经过了一层修饰,是ori的-log10版本
所以上面两者会有差异
然后注意因为是取了-log10之后了,所以padj实际上单调性就反向了
原来padj越小的,小于0.1的,
在-log10之后会越大
也就是说在未过滤文件中筛选的是padj<某一个阈值
到了intra中因为筛选的就是padj>某一个阈值,但是不用担心,在脚本中已经将log10反转过来了;
综上:
参照上述intra脚本,
使用未过滤0.1后的文件group-ori-未filtered,再次进行分析,
时刻注意阈值的使用
必要时修改阈值,用于其他分析
已经做了padj阈值为0.1的分析即默认阈值,后续可以使用0.05或者是0.01的阈值。
后面开始做区室gene的时候:
对于02_Analysis_dchic.Rmd脚本(无论是1vs1,还是_1vs3),
脚本中所统计的是gene symbol作为gene id的数据,但是不同chr有gene的symbol是一致的,所以之前在绘制 upset图的时候才会有交集,都是不严谨的;
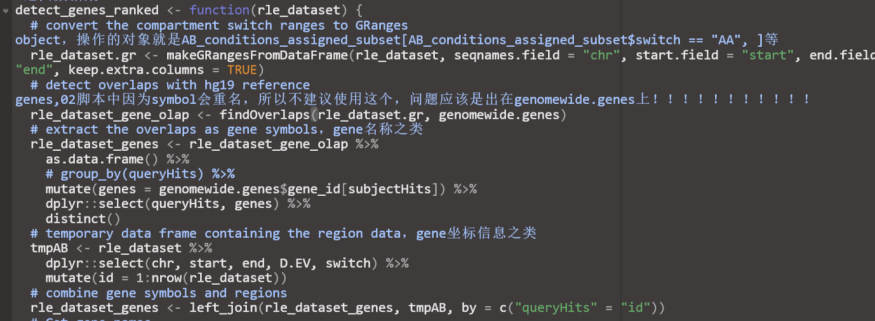
主要是这个使用计算overlap的函数,然后这里修改了一下函数的功能,同样的数据,能够计算出来对应gene id为ensmble的list,
之后如果有需要转换的地方,可以通过bitr函数在不同的gene id之间进行转换;

总之目前:
可以使用原来的symbol的函数,但是不要化upset图,或者是统计交集之类数据
可以在原来的注释数据上将symbol去除重复
当然也可以直接使用上述换算之后的函数ens,在后续需要转换gene id的时候再换算
最后脚本处理得到的汇总信息,依据对应的gene id是ens的还是symbol的
保存如下:
后缀带有ens的就是使用ens的id的,主要是为了后面的upset绘制使用
后面发现其实是不影响的,因为前面的算法还是有问题,没有解决,所以id上尽管是ens还是有重复的
所以还是一切以symbol为主。
之后区室转换的gene联系DEG:

区室转换gene的转录调控信号:TSS富集信号分析(ATAC,K27ac信号等)未做,是否需要交集DEG
Gtf提取区室gene TSS,具体看富集信号的peak文件。
后面做线性回归分析:

DEG的去除,只保留显著区室gene的
注意这里:使用的是全部gene,不是显著与否的区室转换gene,所以这两幅图在padj0.1还是1中的效果是一致的
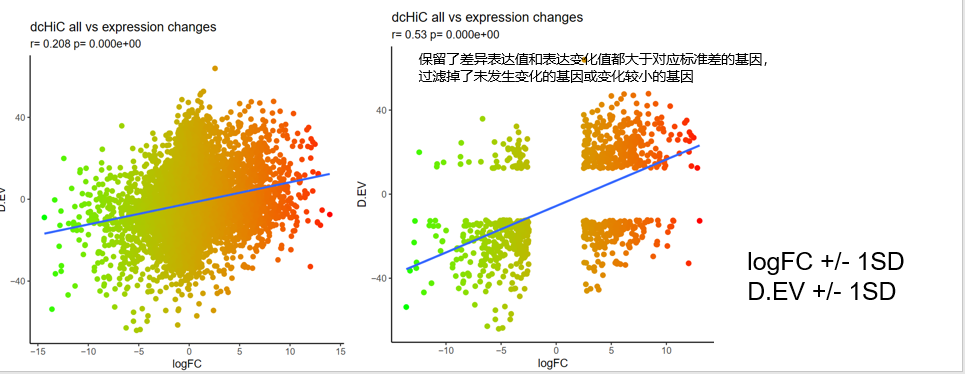
待补充部分:
应该是提取出显著的区室转换gene,然后看在区室转换前后在HMEC以及TNBC中的ATAC,k27ac等信号的差异热图
或者不看gene,直接看这些区域,
获取显著区室转换区域的bed文件:
这里要注意:在之前进行区室分析的时候获取了一个bed文件
然后可以发现其实是有4类型的bed文件混合在一起,
所以可以提取出4类bed文件,然后绘制这些区室转换区域分别在HMEC以及TNBC中的转录调控信号如何,TNBC可以分别绘制或者是使用合并之后的bed文件
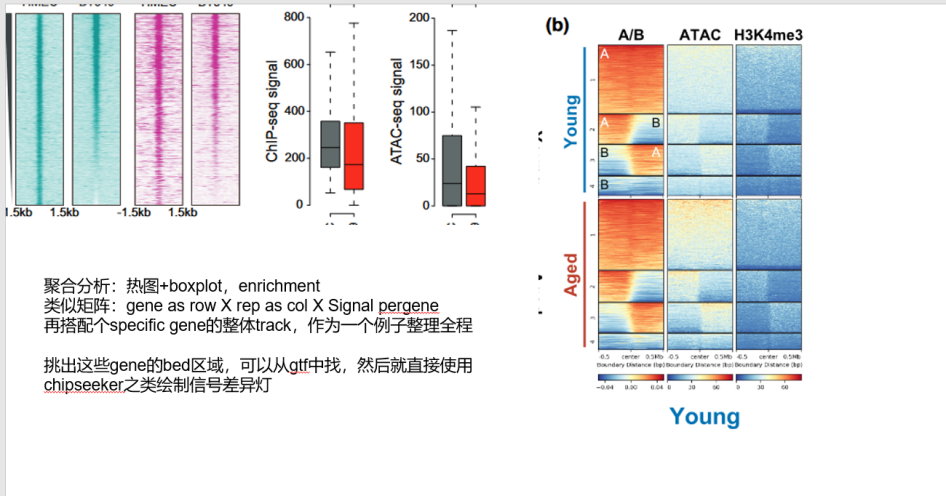

富集分析这一块使用metascape的解释(超几何检验),或者是搞清楚D.EV表征的意思(使用脚本中的GSEA)
至于GSEA的结果先保留barplot+dotplot
以上区室分析部分絮絮叨叨完毕,注意标注为"絮絮叨叨+经验"的部分,仅供参考,不纳入正式分析中
以上部分可以跳过。。。
#4,可以考虑再做拓展的部分:
(1)查看hicexplorer以及GENOVA中对于区室的其余模块分析
(2)子区室sub-compartment的分析:那对照以上compartment都可以再执行一遍
(3)对已有分析模块的深入:
比如说D.EV分布上可以划分比对的样本rep、4类区室转换类型、gene中的up+dnow分面进行处理,不仅仅局限于1个分面
(4)区室+gene的内容可以深入:区室转换区域所overlap的gene,对象是gene就可以做很多模块了,
除了GSEA+GO/KEGG,
还可以搭配其他转录组学相关track(ATAC、H3K27ac的track等),
还可以做WGCNA等,
还可以挖掘TCGA等临床数据库,对gene做生存分析,构建cox模型等(单因子、多因子等),
还可以做免疫浸润分析,或者找到药物靶点的gene做药物基因组分析等;
总之gene有什么模块可以做,这个区室+gene就可以拓展很多
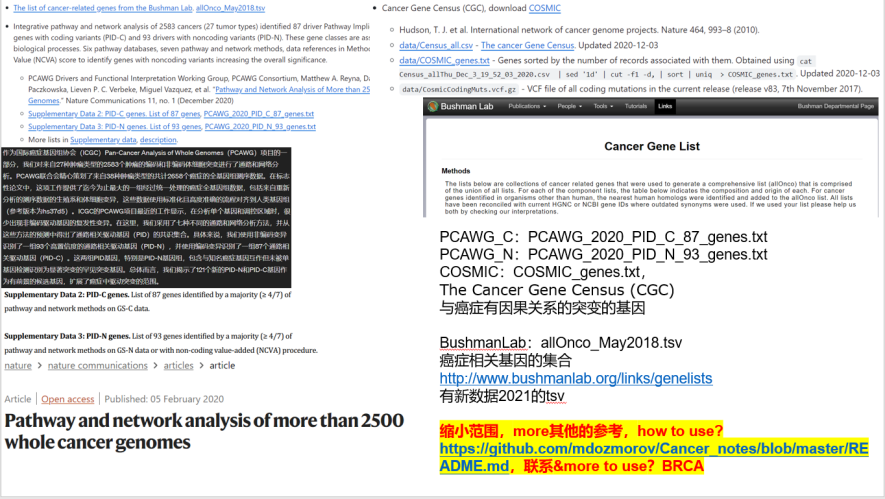
(5)gene还可以做原癌gene注释,主要和癌症研究相关,但是这需要使用一些公共数据库中的cancer标志gene,或者是一些著名lab发表的gene list;
参考脚本02_Analysis_dchic.Rmd

具体这一方面可以深入cancer基因组学+临床生存分析方面,
参考https://github.com/mdozmorov/Cancer_notes/blob/master/README.md,
(6)其他:

可以根据区室gene的bed文件提取对应的TSS位点中的bed,然后使用ATAC或者会死CHIP的peak文件进行chipseeker的注释以及查看信号
总之,再重申一次,直接对照原始脚本+现在处理脚本即可,对照参考注释理解,具体执行还是使用原始脚本,该系列hic系列脚本都这样!
todo:
1,GSEA的区别博客还没有更新
2,但是我们在load_contacts中有2个参数的设置感到困惑:scale_bp以及balancing!
干脆直接问开发者:待解决issuehttps://github.com/robinweide/GENOVA/issues/361