 RNA-seq分析有很多流程,
RNA-seq分析有很多流程,
一般都是上游linux工具获取表达矩阵数据,然后就可以使用下游R包进行处理了,要么是差异DEG表达gene等分析;
因为下游分析其实R包是明确的,毕竟有很多生信分析教程,但是上游的linux如何获取gene表达矩阵,实际上工具就很多了,所以不同套件的工具组合成一个上游分析系列,就会有很多分析方式;
1,上游linux的比对+计数工具选取:
参考https://mp.weixin.qq.com/s/Ny2VYP-M9BxD2V8R5kkXaA
有两种方式比较推荐:

这种方式其实很常见:上游比对/回帖的工具,比如STAR、hisat2+下游计数的featureCounts

直接使用salmon进行定量,但是所谓的"clean fq"是什么,是指经过上游QC的fq文件吗?
另外新增一种方式:
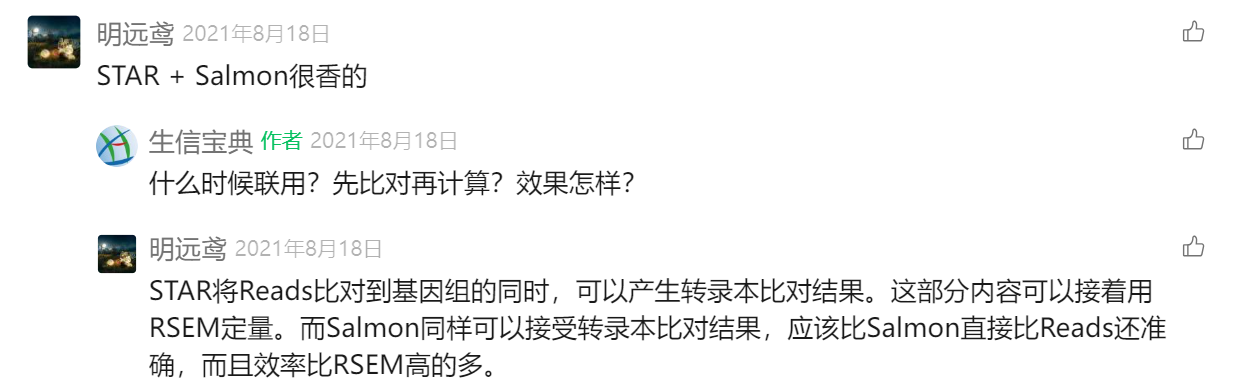

常规QC——》STAR——》Salmon——》edgeR,
主要就是STAR+Salmon是他的linux上游分析工具;
总之例子很多,转录组分析工具是非常的多,可参考https://mp.weixin.qq.com/s/NUEi6oRFL7B3f1FpCD4Xug
下面采用2以及3的方式来处理常规RNA-seq:
(1)Salmon直接上游定量:
参考公众号:https://mp.weixin.qq.com/s/aDxnkK3jhyP6E_wfvza6jQ?search_click_id=11630710159653975537-1726118237844-0581657105
(以及下游的后续CSDN:https://blog.csdn.net/qazplm12_3/article/details/111056012?ops_request_misc=%257B%2522request%255Fid%2522%253A%25222FF4544C-25A8-44B0-8470-554470EF9883%2522%252C%2522scm%2522%253A%252220140713.130102334.pc%255Fblog.%2522%257D&request_id=2FF4544C-25A8-44B0-8470-554470EF9883&biz_id=0&utm_medium=distribute.pc_search_result.none-task-blog-2blogfirst_rank_ecpm_v1~rank_v31_ecpm-3-111056012-null-null.nonecase&utm_term=salmon&spm=1018.2226.3001.4450)
基本等同于https://github.com/nattoh03/Salmon-Quant,
https://github.com/LauYuXuan/salmon_processing/blob/main/sh_salmon.sh,
https://github.com/bissessk/RNA-seq-Analysis-of-Control-and-NRDE2-depleted-Breast-Cancer-Cells/blob/master/1%20-%20Scripts%20and%20Commands/9_salmonQuantifySamples.sh
https://github.com/Alecrim24/Salmon_quant/blob/main/salmon%20on%20all%20reads%20mapped%20to%20the%20h.m%20genome
可以参考https://github.com/SamuelAMiller1/RNA-Seq_Analysis_salmon编写一个全程上游自动化的脚本
首先是下载salmon:
有很多方式,可以直接去salmon官网或者是github官网按照指示,
或者直接mamba search,也可以去bioconda寻找;
#构建索引index,从ucsc浏览器或者ENSEMBL下载参考基因组fa文件、基因注释gtf文件;假设为GRCh38.fa、GRCh38.gtf
#建议使用ensembl,最好是grch38也就是hg38
#这一块整个代码实际上就是参考https://combine-lab.github.io/alevin-tutorial/2019/selective-alignment/# 获取cDNA序列
gffread GRCh38.gtf -g GRCh38.fa -w GRCh38.transcript.fa.tmp# gffread生成的fasta文件同时包含基因名字和转录本名字
grep '>' GRCh38.transcript.fa.tmp | head# 去掉空格后面的字符串,保证cDNA文件中fasta序列的名字简洁,不然后续会出错
cut -f 1 -d ' ' GRCh38.transcript.fa.tmp >GRCh38.transcript.fa# 获取所有基因组序列的名字存储于decoy中
grep '^>' GRCh38.fa | cut -d ' ' -f 1 | sed 's/^>//g' >GRCh38.decoys.txt# 合并cDNA和基因组序列一起,注意:cDNA在前,基因组在后
cat GRCh38.transcript.fa GRCh38.fa >GRCh38_trans_genome.fa# 构建索引 (更慢,结果会更准)
salmon index -t GRCh38_trans_genome.fa -d GRCh38.decoys.txt -i GRCh38.salmon_sa_index

-d的文件(就是上图的decoy)加上整个到index步骤的代码块,都是参考https://combine-lab.github.io/alevin-tutorial/2019/selective-alignment/,也就是上面step1中代码块第3行的参考;


整体上上面大部分的代码是没有问题的,
就是-k参数,也就是k串,默认是31,但是我们在multiqc中实际上是可以知道我们二代测序的reads的长短的,所以可以根据上面这个指示进行修改-k参数,一般是改小。
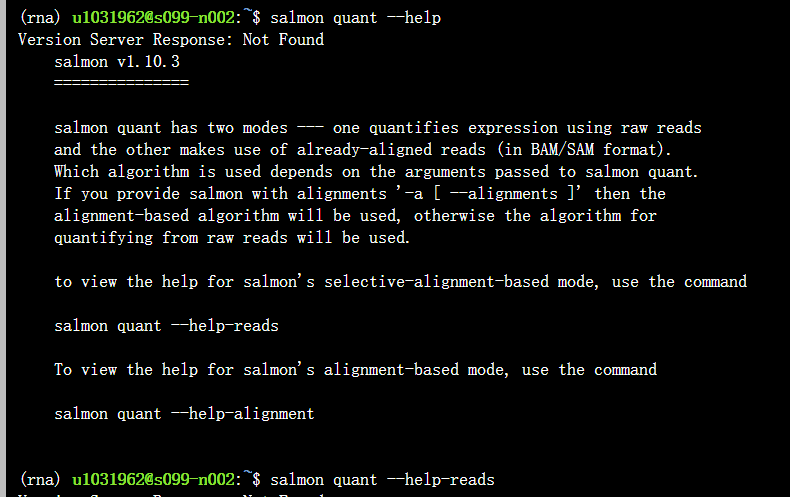
step2:进行定量化,主salmon quant函数
实际执行过程中,参考官网https://salmon.readthedocs.io/en/latest/salmon.html
salmon有两种比对方式,就选其中使用reads的方法此处。因为:
salmon有2个模式mode:
1个是**mapping**-based mode,也就是先index再定量化,即直接对raw reads进行操作的此处(1)Salmon直接定量;

另外1个是alignment-based mode,可以接受STAR的比对/回帖,再Salmon定量化,也就是后面的(2)

因为我们是直接对reads进行定量化,所以此处选用mapping-based mode,

主要使用的参数:
-l A是对于raw reads定量而言的,亦即该模式;
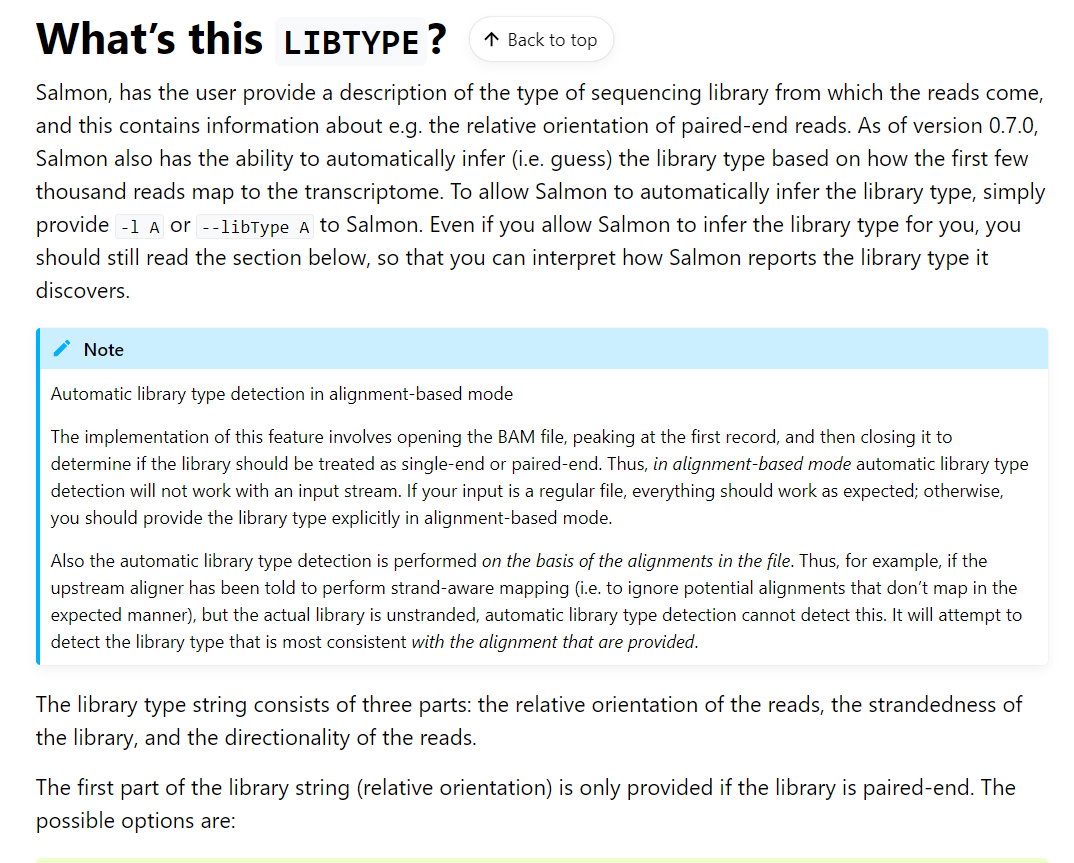
–gcBias 都可以直接使用in any case

-p是线程数

官网推荐的这个参数已经弃用了

-g参数也就是前面step1中提供的gtf文件,只要提取其中的transcript_id以及gene_id两列即可,虽然官网中没有提示这个参数,所以
实际软件与官网提示不一定一致,看issue

其他的参数暂时就没有想到以及使用了,可以查看大多数人的经验缩小参数,然后看看是否必要设置;
当然,直接全部参数都去看才是最保险的!
反正就在这里https://salmon.readthedocs.io/en/latest/salmon.html
注意上游fq单端还是双端会有影响,分别编写了对应shell脚本salmon.sh、salmon_PE.sh、salmon_SE.sh,
**建议先对上游fq文件进行质控QC再执行salmon脚本!然后依据以前对于RNA-seq的处理经验,一般上游multiqc或者是fastqc的质控报告html中并不是所有的red flag error都需要修剪,一般只要trim_galore常规命令修剪,修剪之后依然error的一般不用太在意,因为RNA-seq的reads比较特殊,上游质控可以宽松点;

参考https://github.com/nattoh03/Salmon-Quant
总之常规QC+修剪——>再salmon quant定量化**,所谓的clean fq
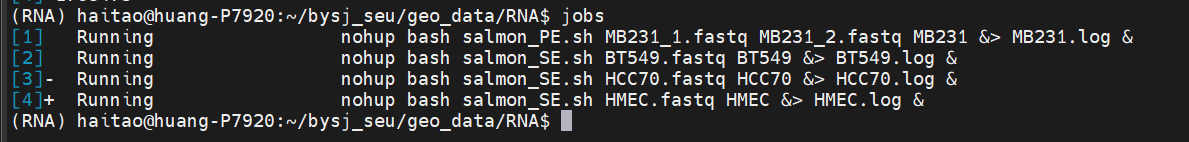
然后输出的记过是quant.xxx.sf文件,

输出格式参考https://salmon.readthedocs.io/en/latest/file_formats.html#fileformats

总体而言,执行过程中官方声明线程数建议在8-12左右,所以我本人是用10线程左右,
总之一句话,对于软件的使用,一定要看官方教程,一定要搞清楚每个参数设置的必要性,不要随便拿别人的脚本来用,很吃亏的!
以上定量化流程并不完善,但是主要函数都列出了,后续再使用时最好对照官网再次进行分析
然后这个sf文件就可以直接开始下游分析了,比如说是DEG分析
综上,该模块整体脚本参考https://github.com/MaybeBio/HiC_pipeline/blob/main/pipeline/hic%20%E5%9F%BA%E7%A1%80pipeline%E9%AA%A8%E6%9E%B6/salmon.sh
(2)Star+Salmon定量:
对于STAR,也和1中一样使用featureCounts,即先STAR再featureCounts,但是也知道STAR自己也能定量化counts,所以为什么需要featureCounts呢、也就是为什么还需要Salmon呢?
参考https://mp.weixin.qq.com/s/OJ1MpHhW08BcZSjMvFs3eg
跑题了,我们这里要做的是先STAR再Salmon。
质控的fastqc+multiqc,以及修剪的trim_galore以及cutadapt在常规脚本中都有,见ChIP/ATAC-seq脚本都有;
参考官方https://salmon.readthedocs.io/en/latest/salmon.html

salmon有2个模式mode:
1个是**mapping**-based mode,也就是先index再定量化,即直接对raw reads进行操作的(1)Salmon直接定量;
另外1个是alignment-based mode,可以接受STAR的比对/回帖,再Salmon定量化,也就是此处的(2)
此法暂略,后续待更新
2,RNA-seq下游分析:
(1)DEG分析+GSEA:
原始参考脚本PDXHiC_supplemental/Fig2_RNA-seq/01以及02脚本,
修改见https://github.com/MaybeBio/TNBC-project/tree/main/2%2CCompartment同名,
调整之后参考见HiC_pipeline/pipeline/hic 基础pipeline骨架/01_TPMCalc_DataPreproc.R以及HiC_pipeline/pipeline/hic 基础pipeline骨架/02_DEG_Analysis_edgeR.Rmd,
可参考https://mp.weixin.qq.com/s/bXv1KG-p307pZ5i94AYnTQ,
主要就是使用salmon中的输出文件quant.sf文件,注意是转录本定量的文件,不是gene的!
对应脚本中也有这一步骤,注意将注释文件对应好即可,


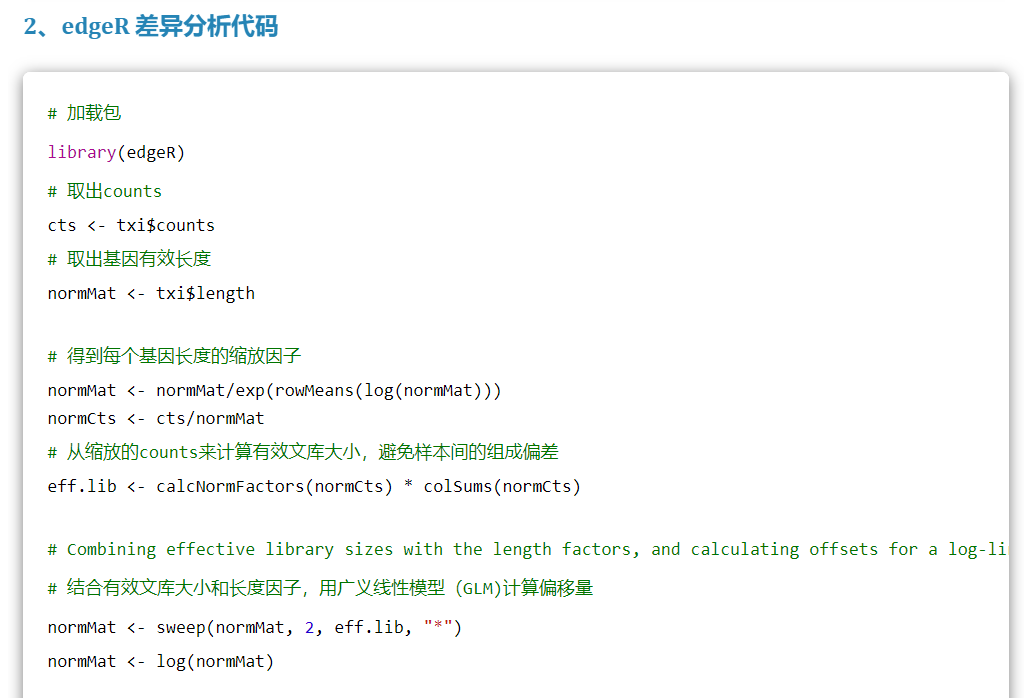
总之是可以参考
https://mp.weixin.qq.com/s/bXv1KG-p307pZ5i94AYnTQ,也可以参考自己脚本中上游salmon输出到edgeR处理DEG;
DEG处理之后就是功能富集分析,即GSEA,可以使用clusterprofiler包,因为有logFC等rank数据,所以可以使用GSEA,clusterprofiler绘制出来的图已经足够了;
注意GSEA与常规的GO/KEGG中的过表达分析不同,在算法上不同,使用参考不同,前者rank,后者超几何test,可以参考博客,待更新。。。
(2)其他:
Fig2_RNA-seq/

3,其余pipeline,RNA-seq相关
很多script,可以参考仓库RNA-seq



![[数据集][目标检测]车窗状态检测车窗开关检测数据集VOC+YOLO格式299张3类别](https://i-blog.csdnimg.cn/direct/614e46e418304c09b7f0b77d47496086.png)