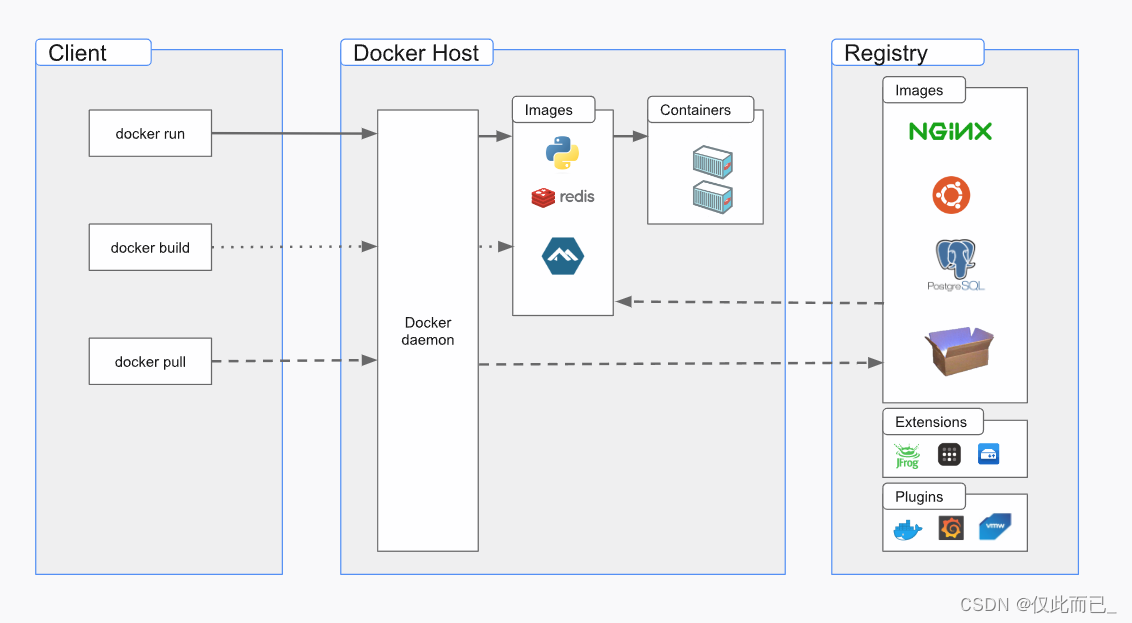
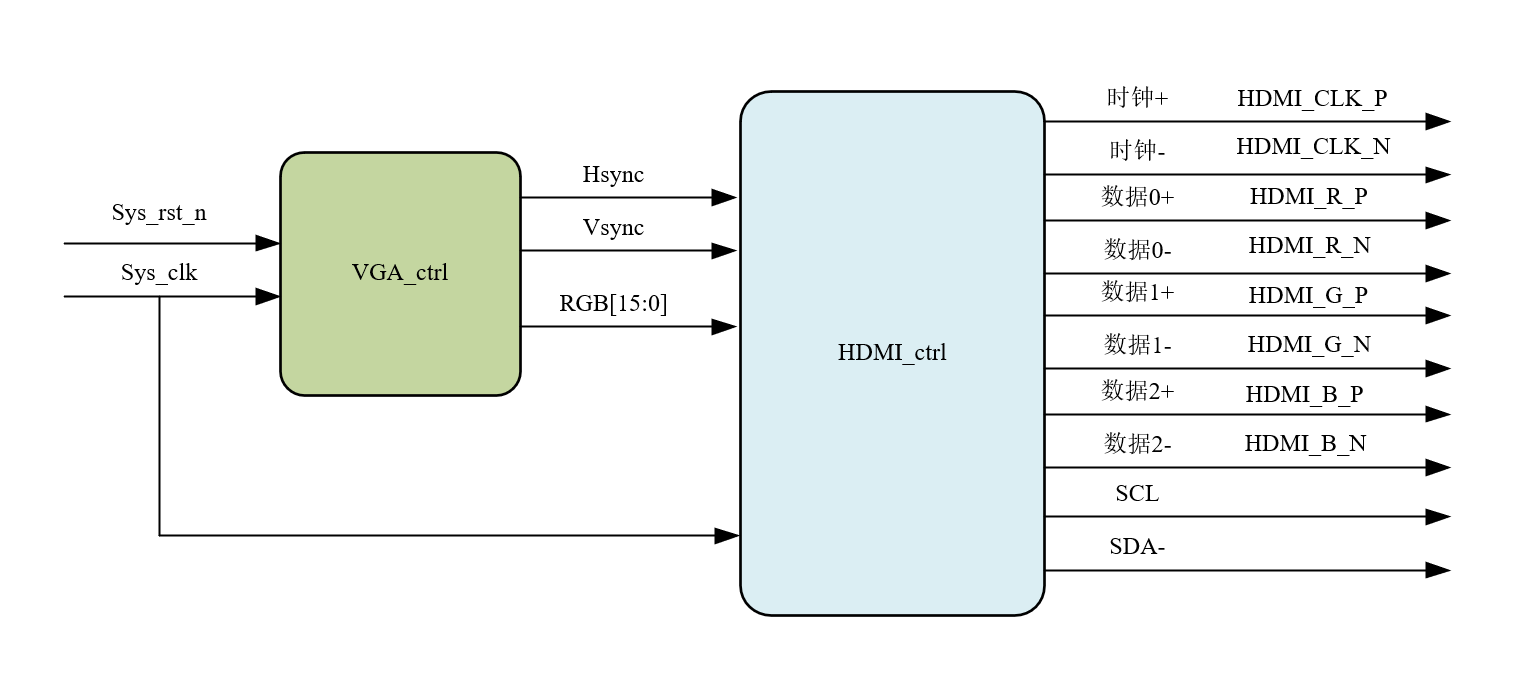
文章标题:sAMP-PFPDeep: Improving accuracy of short antimicrobial peptides prediction using three different sequence encodings and deep neural networks
代码:https://github.com/WaqarHusain/sAMP-PFPDeep
一、问题
短抗菌肽(sAMPs):红色是α-helices,蓝色为随机coil

最著名的生物计算问题之一是在离散模型中描述生物序列,使其关键序列特征不被改变。以载体形式表达生物序列可能导致失去其重要的基于序列的特征。除此之外,各种基于物理化学特征的方法,主要包括氨基酸组成(AAC)、伪氨基酸组成(PseAAC)、归一化氨基酸组成(NAAC)、疏水性、净电荷、等电点、α-螺旋倾向、β-片倾向和转向倾向,已经被提出用于预测amp,这些表征具有很强的预测肽序列性质的能力。
将序列转换为图像时,分别考虑了包含位置、频率和12个理化特征之和信息的三个通道。
二、Materials and methods
预测sAMPs,即具有少于或等于30个氨基酸残基的序列长度的肽。流程:

1、Training and benchmark dataset
本研究使用了先前数据集【Deep-AmPEP30: improve short antimicrobial peptides prediction with deep learning】。数据集由1529个sAMPs和1529个Non-sAMPs组成,表明数据集是平衡的。
数据样本:

最终用于训练的数据集包含1529 + 1529 = 3058个样本。数据集已经经过CDHIT处理,去除冗余的阈值为0.8,即排除相似性超过80%的序列。由于已经执行该预处理,因此在本研究中没有重复该步骤,并且数据集被用作训练目的。188个多肽的基准数据集,包括来自同一研究的94个sAMPs和94个非sAMPs。
2、Sequence to image generation
Sequence to square matrix conversion
将序列转换为方阵。首先,在所有序列中填充假氨基酸,即序列长度小于30的X。这有助于使数据集中的所有样本具有均匀的长度。在下一阶段,将这些序列转换为5 × 6矩阵,例如:

Square matrix to 3-channel image conversion
方阵被转换为3通道图像。为此,对每个通道进行了不同的计算。
第一通道:编码矩阵。每个氨基酸的编码从1到20,X氨基酸被认为是零。
第二通道:各自序列的方阵中的氨基酸被替换为氨基酸频率。例如,如果氨基酸A,即丙氨酸在一个序列中出现3次,则该序列中的每个A都被替换为3。频率矩阵。频率分布:
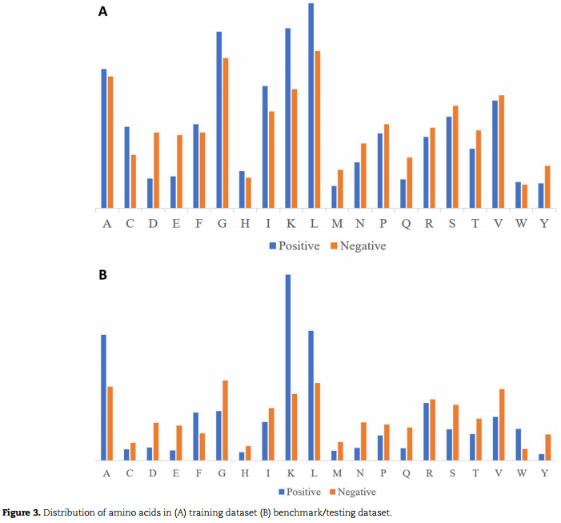
第三通道:氨基酸的理化特性。PepData从CRASP程序中收集了属性值,除Solvent_Exposed_Area,而Solvent_Exposed_Area的值取自(http://prowl.rockefeller.edu/aainfo/access.htm)。

在将所有三个通道划分为单个图像之前,所有通道都在0-255的范围内归一化。这有助于生成三个实际的均匀通道,并在合并它们后,从每个肽序列生成一个3通道图像。
3、Classification through VGG-16 and RESNET-50
经过20个epoch后,模型收敛。为优化所有参数,使用Grid进行超参数调优。VGG-16和RESNET-50:

两种神经网络的最小输入层尺寸均为32 × 32 × 3,而本研究生成的图像为5 × 6 × 3。因此,为将这些微小的图像传递给模型,对图像执行零填充。
4、Validation study
为验证,采用基于分子对接(AutoDock Tools和AutoDock Vina)。首先,建立从UniProt中检索长度≤30个残基的AMP的数据集。通过关键字抗菌[KW-0929]进行检索,长度设置为∗TO30,检索到728个已审查的肽序列。随后,为去除检索序列中的冗余,应用CD-HIT,相似度阈值为60%,从原始的728个序列中检索到301条肽序列。
除预测标签外,对于阳性样本,还计算以p值(概率)表示的预测分数,因为这些收集的肽实际上都是阳性的。这些肽被归类为阳性sAMPs,进行三级结构预测,并使用SWISSModel建模。通过与八种已知细菌受体的分子对接,评估这些肽的抗菌潜力。

每次对接后计算反应的结合能,并利用这些结合能计算μM中的抑制常数Ki值为:

其中G为结合能,T为温度,为298.15 K, R为气体常数,为1.9872036 kcal/mol。
在进行分子对接时,使用AutoDock Tools为观察到的每个蛋白质的结合位点生成一个Grid box dimensions(size),并记录。
使用AutoDock Vina进行分子对接,并计算所有对接肽的结合亲和力值,以了解它们与感兴趣的蛋白质的相互作用。
在本研究中,采用E = 4、E = 8、E = 16、E = 32、E = 64和E = 128六种不同穷举启发式的对接仿真方法。然而,在穷举E = 8后,结合方面未见改善,因此,报告E = 8的结果。
为更好地描述,预测分数,即p值与所有肽的结合能(γG)和抑制常数(Ki)相关。
5、Evaluation of performance

三、Results and discussion
1、Estimation of training performance
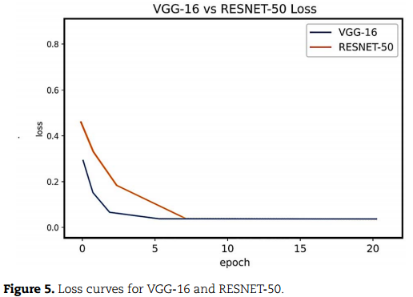
VGG-16的训练效果优于ResNet-50:

基于VGG-16的预测产生了1502个真阳性和1504个真阴性,假阳性和假阴性分别为25个和27个。预测1484个真阳性和1456个真阴性,而假阳性和假阴性分别为73和45。这表明VGG-16的精度与RESNET-50相比有显著差异:

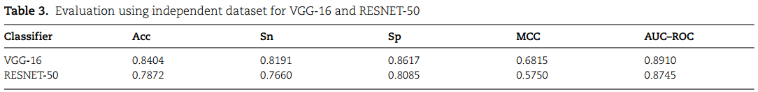
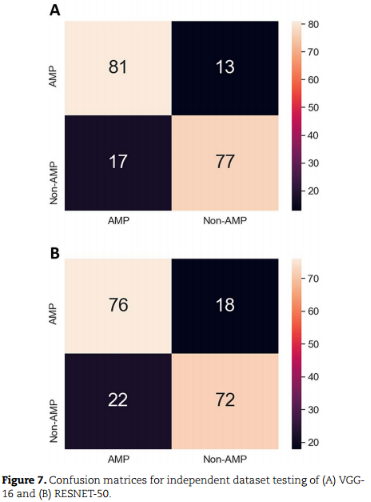
2、Evaluation of predictors based on independent dataset testing
使用了94个samp和94个非samp的未见数据。VGG16在所有评估指标方面都比RESNET-50表现出更好的结果:
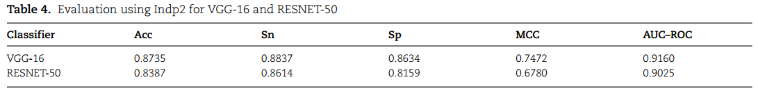
另一个独立的数据集,Indp2,包括1032个samp和1032个非samp,仅考虑长度在11 ~ 30个残基之间的序列,用于测试模型:

3、Comparative analysis with state-of-the-art methods

4、Validation through molecular docking
为描述sAMP-PFPDeep预测与对接结果的相关性,绘制结合能(γG)与预测评分(p值)的相关图:
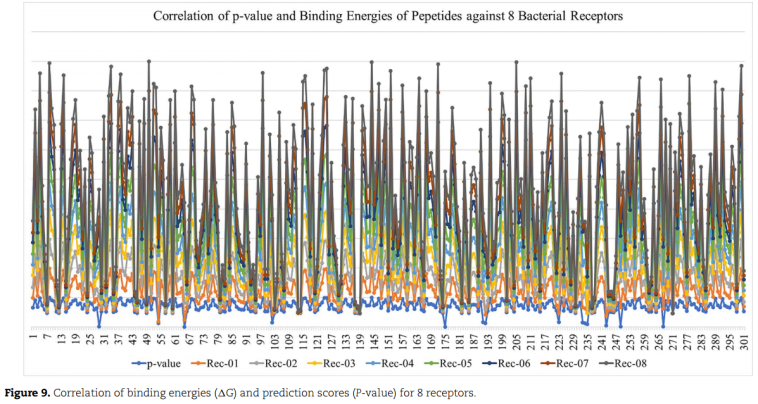
预测结果与图中趋势线所示的结合能密切相关,除了少数被错误预测为阴性的肽(non-sAMPs)。趋势线的起伏对所有287个肽都是同步的。此外,这些肽与8种细菌受体的结合能较高,表明它们具有较强的抗菌活性候选性,而sAMP- pfpdeep对sAMP的预测也证明了这一点。这表明,通过提出的方法预测为sAMP的肽是对细菌受体表现出强结合能的候选肽。此外,该方法主要用途是,实验生物学家可以在进行分子对接模拟或任何体外实验之前,通过所提出的方法预测肽的类别是sAMPs还是non-sAMPs。