前列腺癌(PC)是全球第二大最常见的男性癌症,每年估计有375,304人死亡。虽然雄激素剥夺疗法(ADT)仍然是晚期前列腺癌的当前标准治疗方法,但大多数患者最终进展并发展为致命的转移性去势抵抗性前列腺癌(mCRPC)。
PTEN(一种抑癌基因)功能丧失发生在大约50%的mCRPC患者中,并与不良预后、治疗结果和对免疫检查点抑制剂的抵抗有关。最近的临床研究表明,抑制PI3K/AKT双通路和阻断雄激素轴可以轻微改善PTEN缺陷的mCRPC患者的无进展生存率,但这种有限疗效的机制尚不清楚。
为了阐明潜在的耐药机制,2023年5月15日,美国芝加哥大学医学系的Akash Patnaik研究团队在Clinical Cancer Research(IF:11.5)杂志上发表了题为“Reversal of lactate and PD-1-mediated macrophage immunosuppression controls growth of PTEN/p53-deficient prostate cancer”的研究论文,该研究在前列腺特异性PTEN/P53基因工程小鼠模型中进行了联合临床试验,结果表明PI3Ki和aPD1分别逆转乳酸和PD-1介导的TAM免疫抑制,与ADT联用可控制肿瘤生长,值得在PTEN/P53缺陷的mCRPC患者中开展进一步的临床研究。

发表单位:美国芝加哥大学医学系
期 刊:Clinical Cancer Research(IF:11.5)
发表日期:2023年5月15日
研究技术:scRNA-seq和RNA-seq(爱基百客均可提供)
一 材料方法
前列腺特异性PTEN/p53缺陷(Pb-Cre; PTENfl/fl Trp53fl/fl)小鼠在16周龄时通过超声波筛查自身前列腺肿瘤发生情况。在实体瘤形成后(超声成像下实体瘤的长轴直径达到5mm时),小鼠接受 degarelix(0.625 mg/只小鼠,每 28 天一次,ADT 组)、copanlisib(14 mg/kg,每隔一天一次)、PD-1 抗体(aPD-1,200 μg//只小鼠,每隔一天一次)单药或它们的组合药处理。
二 研究思路
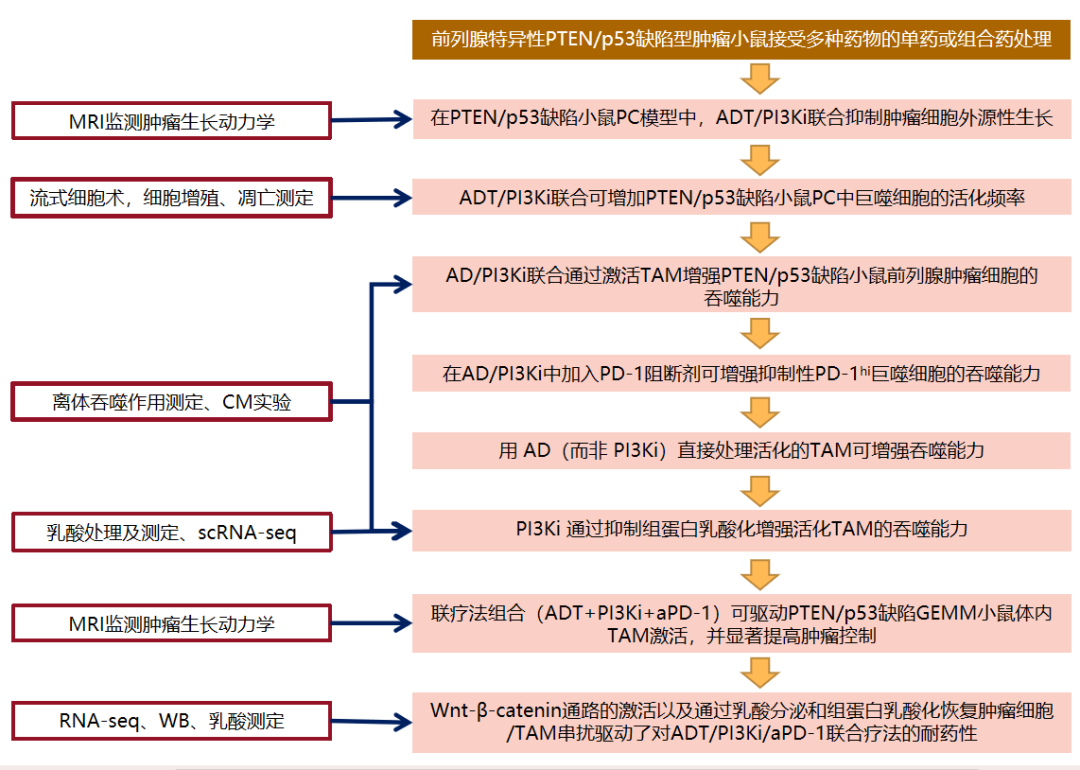
三 研究结果
01 在PTEN/p53缺陷小鼠PC模型中,ADT/PI3Ki联合抑制肿瘤细胞外源性生长
先前研究表明,PTEN缺陷型AVPC患者的ADT反应性有限。作为阐明ADT与PI3Ki在PTEN/p53缺陷小鼠PC中的联合作用的第一步,16-20周龄Pb-Cre; PTENfl/fl Trp53fl/fl小鼠(已建立了100- 150mm3实体瘤)用degarelix(促黄体生成素释放激素(LHRH)拮抗剂,化学阉割),单独或联合copanlisib(泛PI3K抑制剂)处理4周。采用磁共振成像(MRI)监测肿瘤生长情况。
虽然ADT显著降低了所有小鼠的血清睾酮水平至去势水平,但客观缓解率(ORR)为16.7%,表明大多数小鼠对ADT产生了原发性耐药(图1A)。与未处理对照相比,Copanlisib无论是单独使用还是与ADT联合使用,都能完全抑制PI3K通路,ORR分别增加了37.5%和25%(图1A),从而证实了已发表的临床试验数据。特别是,相对于单一药物(0%),degarelix/copanlisib联合处理观察到肿瘤缩小(12.5%),这表明两者之间存在协同作用。相比之下,在ADT/copanlisib联合处理7天后收获的肿瘤提取物显示,相对于单一药物和未处理的对照组,TME中Ki67+肿瘤细胞(流式细胞术测量的增殖标志物)减少了2.3倍(图1B)。这些发现提示ADT/PI3Ki联合在PTEN/p53缺失的前列腺GEMM中通过肿瘤细胞外源性机制发挥其抗癌活性。

图1 通过增加肿瘤相关巨噬细胞(TAM)在TME内的浸润/激活,ADT和PI3Ki联合疗法在PTEN/p53缺陷小鼠PC中显示出部分抗肿瘤反应。
02 ADT/PI3Ki联合可增加PTEN/p53缺陷小鼠PC中活化巨噬细胞的频率
为了阐明ADT/PI3Ki联合疗法在TME内的抗癌机制,作者对Pb-Cre; PTENfl/fl Trp53fl/fl小鼠在degarelix单独或与copanlisib联合处理7天后的前列腺肿瘤进行了免疫分析。未经处理的PTEN/p53缺陷前列腺肿瘤的TME中富含肿瘤相关巨噬细胞(TAM,占总CD45+细胞的25%)和粒细胞-髓系衍生抑制细胞(Gr-MDSC,占总CD45+细胞的38%),T细胞相对缺乏(CD4表达占总CD45+细胞的3%,CD8表达,占总CD45+细胞的1%)(图1C)。
作者观察到,与未处理的对照组相比,在接受degarelix处理的PTEN/p53缺陷瘤小鼠中,总TAM的频率增加了1.4倍,而同时服用copanlisib并没有增强这种情况(图1C)。此外,相对于未处理的对照组和单独的copanlisib, degarelix/copanlisibli处理导致表达PD-1的TAM显著增加(图1D)。重要的是,copanlisib处理导致活化(MHC-IIhi) TAM频率增加2.3倍,与未处理对照相比,与ADT联合处理显著增强至3.6倍(图1E)。综上所述,这些数据表明,相对于相应的单一药物,ADT/PI3Ki联合使用显著增强了TME内TAM的激活。
由于ADT/PI3Ki联合处理相对于未处理的对照组增加了MHC-II和PD-1表达TAM的频率,作者重点研究了MHC-IIhi/lo和PD-1hi/lo TAM亚群在肿瘤生长控制中的作用,发现活化的TAM (MHC-IIhi/PD-1lo和MHC-IIhi/PD-1hi)与ORR呈正相关,而免疫抑制的MHC-IIlo/PD-1lo和MHC-IIlo/PD-1hi TAM与ORR呈负相关(图1F-G)。总的来说,这些数据表明TAM活化与抗肿瘤反应密切相关,而抗肿瘤反应被TAM内PD-1的表达减弱。
03 AD/PI3Ki联合通过激活TAM增强PTEN/p53缺陷小鼠前列腺肿瘤细胞的吞噬能力
为阐明活化(MHC-IIhi/PD-1lo和MHC-IIhi/PD-1hi)TAM控制Pb-Cre; PTENfl/fl Trp53fl/fl小鼠体内的肿瘤生长的机制,作者研究了copanlisib与AD(模拟体内degarelix处理)相结合对TAM介导的PTEN/p53缺陷GEMM-肿瘤衍生PC细胞系AC1和SC1的吞噬作用的影响。作者首先进行了共培养实验,以确定分离的TAM亚群(MHCIIhi/lo和PD-1hi/lo TAM)与AC1/SC1细胞在基线条件下的相对吞噬活性(图2A)。与TAM亚群/ORR相关数据一致(图1G),活化的MHC-IIhi/PD-1lo TAM表现出最高的吞噬活性,与MHC-IIhi/PD-1hi TAM相比,AC1/SC1细胞的摄取增加了14.5倍。相比之下,与MHC-IIlo/PD-1hi TAM相比,MHC-IIhi/PD-1hi和MHC-IIlo/PD-1lo TAM亚群对AC1/SC1细胞的吞噬能力分别增加了8.5倍和2.5倍(图2B)。重要的是,激活的(MHC-IIhi) TAM与失活的(MHC-IIhi) TAM群体相比,PD-L1和CD206的表达水平显著降低,与PD-1状态无关。此外,当与AC1或SC1细胞共培养时,与失活的MHC-IIlo/PD-1hi TAM相比,CD206在失活的MHC-IIlo/PD-1hi TAM中的表达更高(图2C和D)。总的来说,这些数据表明,TAM中的PD-1表达增强了它们的免疫抑制表型,抑制了它们的吞噬活性,而它们的活化状态无关。

图2 PD-1上调抑制活化的TAM的吞噬能力。
接下来,作者研究了AD单独或与copanlisib联合用药对体外AC1和SC1细胞吞噬功能的影响,涉及上述4个TAM亚群。虽然AD导致MHC-IIhi/PD-1lo TAM对AC1和SC1细胞的吞噬作用显著增加(分别为19.6倍和3.8倍),但与未处理组相比,AD并未改变MHC-IIhi/PD-1hi TAM的吞噬活性(图3B和C),这表明PD-1表达对ADT诱导的吞噬作用有抑制作用。相比之下,单药copanlisib处理显示MHC-IIhi/PD-1lo TAM(分别为11倍和3倍)和MHC-IIhi/PD-1hi TAM(分别为12倍和7倍)对AC1和SC1细胞的吞噬作用增加。重要的是,与未处理的对照组相比,AD/copanlisib联合处理导致MHC-IIhi/PD-1lo TAM对AC1和SC1细胞的吞噬能力分别增加32倍和9倍。与未处理的对照相比,联合用药还导致MHC-IIhi/PD-1hi TAM对AC1和SC1细胞的吞噬能力分别增加20.4倍和11.8倍(图3B和C)。与未处理对照相比,AD/copanlisib联合用药或相应的单药处理均未改变MHC-IIhi/PD-1lo或MHCIIlo/PD-1hi TAM的吞噬活性(图3B和C)。总的来说,这些数据表明,AD和PI3Ki对活化的TAM亚群及其对PTEN/p53缺陷前列腺肿瘤细胞的吞噬作用有不同的影响,相对于单药,联合用药的效果更强。此外,AD/PI3Ki介导的吞噬作用在失活的TAM亚群中被显著抑制。

图3 ADT/PI3Ki联合处理可通过激活TAM显著增强PTEN/p53缺陷小鼠癌细胞的吞噬功能。
04 在AD/PI3Ki中加入PD-1阻断剂可增强抑制性PD-1hi巨噬细胞的吞噬能力
鉴于作者的发现,基线和AD/ pi3ki诱导的PTEN/p53缺陷的GEMM肿瘤衍生细胞的吞噬能力被激活的TAM亚群中PD-1的高表达所减弱(图2B和3B),作者接下来评估了aPD-1单独或与copanlisib联合,对TAM介导的AC1/SC1细胞吞噬能力的影响,无论有或没有AD(图4A)。作者观察到,与AD/Copanlisib联合处理(分别为2.7倍和2.1倍)、APD-1单一处理(分别为9.3倍和3.9倍)和未经处理的对照组(分别为28倍和11.7倍)相比,经AD/Copanlisib/APD-1联合处理后,荧光激活细胞分选(FACS)分离的抑制性MHC-IIlo/PD-1hi TAM对AC1/SC1细胞的吞噬活性显著增加(图4B和图4C)。与未处理的对照组相比,AD/copanlisib/aPD-1联合处理后,抑制性MHC-IIlo/PD-1hi的TAM上MHC-II表达增加,表明AD/copanlisib/aPD-1联合处理后TAM重编程。鉴于MHC-IIhi/PD-1hi在TAM亚群中的基线吞噬活性较高,与MHC-IIlo/PD-1hi亚群相比,加入aPD-1的所带来的相对增加幅度较小,如AD/Copanlisib/APD-1组合显示AC1/SC1细胞的吞噬功能部分增加(分别为AD/Copanlisib组合的1.4倍和1.1倍)、aPD-1单一处理组(分别为12.8倍和4.8倍)和未经处理的对照组(分别为32倍和12倍,图4B和C)。这些数据表明,在抑制性PD-1hi TAM中阻断PD-1可以克服PD-1对PTEN/p53缺陷TME中肿瘤细胞吞噬功能的免疫抑制作用。
与AD对照相比,添加aPD-1与AD联合导致MHC-IIhi/PD-1hi TAM亚群对AC1和SC1细胞的吞噬能力分别增加7.4倍和5.7倍。有趣的是,与单独AD相比,AD/aPD-1联合不足以增强MHC-IIlo/PD-1hi TAM的吞噬作用,这可能与这种免疫抑制性TAM亚群中更高的激活阈值有关(图4B和C)。相对于copanlisib单一疗法,copanlisib/aPD-1不增加MHCIIhi/PD-1hi和MHC-IIlo/PD-1hi TAM对AC 1或SC 1细胞的吞噬活性(图4 B和C)。总之,这些数据证明向AD或AD/copanlisib组合中添加aPD-1增强了PD-1hi激活的TAM的吞噬活性。
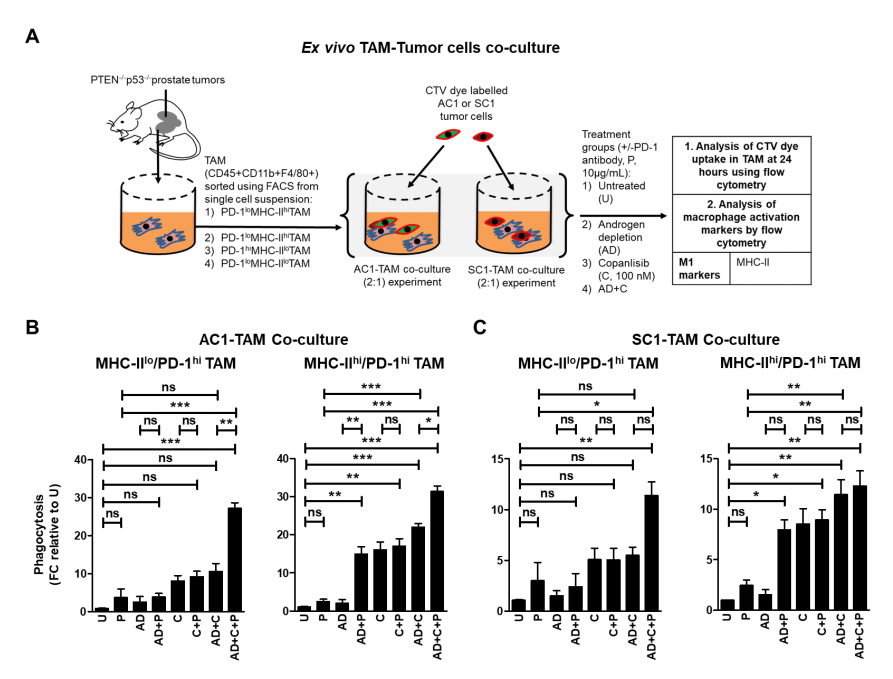
图4 在雄激素耗竭/PI3Ki中添加PD-1阻断可增强抑制性PD-1hi巨噬细胞的吞噬能力。
05 用 AD(而非 PI3Ki)直接处理活化的 TAM 可增强吞噬能力
为了了解AD如何影响TAM的功能,作者用AD、PI3Ki或它们的组合对FACS分选的TAM进行预处理,然后使用PTEN/p53缺陷型GEMM肿瘤来源的AC1和SC1细胞作为靶群体进行吞噬实验(图5A)。有趣的是,相对于未处理的对照,AD显示MHC-IIhi/PD-1lo TAM的吞噬能力增加6.5倍,而同时使用copanlisib和/或aPD 1阻断并不会增强这种能力。关键地是,MHC-IIhi/PD-1hi TAM需要添加aPD-1才能实现类似的吞噬诱导(图5 B和C)。AD/aPD-1联合并没有改变MHC-IIlo/PD-1lo TAM和MHC-IIlo/PD-1hi TAM的吞噬活性。与观察到的吞噬数据(图5B和图5C)一致,AD直接激活TAM,而同时使用copanlisib或aPD-1不会加剧这种情况。综上所述,这些数据表明,AD通过增加TAM的总浸润和MHC-IIhi/PD-1lo TAM亚群的激活/吞噬活性,促进PTEN/p53缺陷PC的抗肿瘤免疫反应,而MHC-IIhi/PD-1hi激活的TAM亚群的吞噬能力增强需要同时阻断PD-1。

图5 单独用雄激素消耗和与aPD-1组合对TAM进行离体预处理,可分别增加MHC-IIhi/PD-1lo和MHC-IIhi/PD-1hi TAM 亚群对 PTEN/p53缺陷GEMM肿瘤细胞的吞噬作用。
06 PI3Ki 通过抑制组蛋白乳酸化增强活化 TAM 的吞噬能力
由于直接使用copanlisib并未直接改变活化TAM的激活状态和吞噬能力(图5B-C),作者推测copanlisib 通过克服 PTEN/p53 缺陷的 PC细胞的免疫抑制分泌组,间接增强 TAM 的激活/吞噬能力,从而促进抗癌免疫。为了验证这一假设,作者进行了体外条件培养基(CM)实验(图6A),观察到活化的MHC-IIhi/PD-1lo和MHCIIhi/PD-1hi TAM的吞噬能力增加了4.5倍(图6B)。由于PI3K通路是癌细胞葡萄糖代谢和乳酸产生的关键驱动因素,作者接下来评估了copanlisib对体内外 CM中乳酸释放的影响。作者观察到,相对于未经处理的对照组,乳酸水平在copanlisib作用下下降了18.1%。与这一发现相印证的是,作者观察到 AC1/SC1 细胞对copanlisib的反应中也有类似的乳酸产生减少。
鉴于最近的研究结果,组蛋白乳酸化(H3K18la)可以驱动TAM内的免疫抑制,作者对体外CM中乳酸含量的减少伴随着活化的TAM内组蛋白乳酸化的减少的假设进行了验证。作者观察到H3K18la在MHC-IIhi/PD-1lo和MHC-IIhi/PD-1hi TAM亚群中明显减少(图6C),这表明PI3Ki诱导的肿瘤细胞内乳酸抑制和活化TAM内组蛋白乳酸化减少增强了肿瘤细胞的吞噬能力。为了明确这一假设,作者测试了在体外CM中添加外源性乳酸对活化TAM吞噬能力的影响(图6A)。有趣的是,作者观察到通过激活MHC-IIhi/PD-1lo和MHC-IIhi/PD-1hi TAM(图6B),可消除copanlisib诱导的AC1/SC1细胞吞噬增强作用(图6B),同时这些TAM亚群中组蛋白乳酸化也得到了恢复(图6 C)。重要的是,用 copanlisib 直接处理不会改变 TAM 的组蛋白乳酸化状态。总之,这些数据表明,copanlisib处理减少了PTEN/p53缺陷PC细胞的乳酸生成和MHC-II+ TAM亚群中的继发性组蛋白乳酸化,从而增强TME内TAM的活化/吞噬作用。
作者的临床前研究结果表明,肿瘤细胞的有氧糖酵解活性(即肿瘤细胞产生乳酸)与晚期人类PC的巨噬细胞吞噬/激活之间存在机理联系,为了验证这一研究结果,作者对骨转移 PC 患者样本BMET-1和BMET-2进行了scRNA-seq。作者观察到癌细胞中明显更高的有氧糖酵解活性,BMET-2比BMET-1对TAM吞噬的抑制相应更高(图6D)。同时,作者还对mCRPC患者转移淋巴结标本(LMET-1、-2和-3)进行了scRNA-seq检测。虽然TAM的数量不足以进行吞噬基因特征分析,但作者观察到肿瘤细胞内的有氧糖酵解活性与髓系细胞内的M1极化状态之间存在负相关(图6E),从而验证了肿瘤细胞内在糖酵解在转移性PC微环境中逃避TAM介导的天然免疫吞噬反应中的作用。

图6 PI3Ki可抑制经处理的 PTEN/p53缺陷 PC 细胞产生乳酸,并抑制活化的 TAM 内组蛋白乳酸化,从而增强 TAM 的吞噬能力。
07 ADT/PI3Ki/aPD-1诱导的体内巨噬细胞活化可控制60%的Pb-Cre; PTENfl/fl Trp53fl/fl小鼠的肿瘤生长
基于作者的发现,ADT、aPD-1和 PI3Ki处理可通过不同机制在4个TAM亚群克服 TAM的免疫抑制,作者确定了ADT/PI3Ki/aPD-1组合是否会增强 Pb-Cre; PTENfl/fl Trp53fl/fl小鼠的抗癌反应,相对于单一和双重对照组。作者用aPD-1单独或与copanlisib和ADT联合处理前列腺荷瘤小鼠,并使用MRI测量肿瘤生长动力学。在ADT中添加aPD-1后,28天的ORR提高到33.3%(相比之下,单独ADT为16.7%)(图7A)。与单独添加copanlisib(37.5%)相比,在copanlisib中添加aPD-1没有观察到ORR的增加(37.5%)(图7A),这反映了作者共培养的吞噬数据(图4B和C)。重要的是,用ADT/copanlisib/aPD-1联合疗法处理的小鼠,到28天时,相对于单药和双药对照组,ORR显著增加了60%(图7A、1A),这与作者的体内外吞噬数据相关(图4B和C)。这种 ORR 伴随着 TME 中活化 TAM 频率的增加(图7B)。重要的是,氯屈膦酸钠联合处理可消耗活化的(MHC-IIhi/PD-1lo 和 MHC-IIhi/PD-1hi)TAM,完全消除了ADT/copanlisib/aPD-1 组合诱导的肿瘤控制(图7A-B )。此外,细胞因子阵列分析显示,在ADT + copanlisib + aPD-1处理后,肿瘤单细胞悬液中的促炎介质(IL-1α、TNFα、CCL-22、CCL-5、IL-6和CXCL-16)增加,而抗炎细胞因子(M-CSF和CCL-6)减少,这支持了作者的主要假设,即活化/极化的巨噬细胞在很大程度上是三联疗法引起的抗肿瘤反应的原因。总之,这些数据证明,相对于 ADT/PI3Ki 组合,三联疗法组合(ADT+PI3Ki+aPD-1)可驱动PTEN/p53缺陷GEMM小鼠体内TAM激活,并显著提高肿瘤控制。
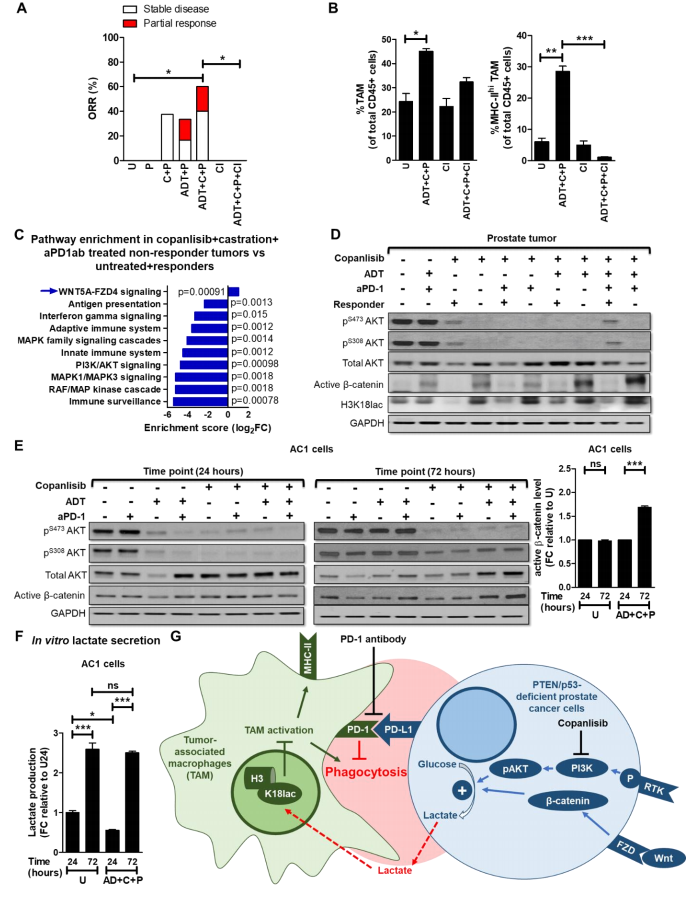
图7 ADT + PI3Ki + aPD-1抗体通过激活TAM导致PTEN/p53缺陷GEMM的肿瘤控制,在无应答者中观察到反馈Wnt/β-catenin激活介导的乳酸和组蛋白乳酸化恢复。
08 在 ADT/PI3Ki/aPD-1联合处理后的无应答 Pb-Cre; PTENfl/fl Trp53fl/fl小鼠中观察到的 Wnt/β-catenin信号的反馈激活,可恢复乳酸介导的组蛋白乳酸化(H3K18la)并抑制巨噬细胞的吞噬作用
为了阐明无应答小鼠对ADT/PI3Ki/aPD-1联合疗法的耐药机制,对无应答小鼠的肿瘤进行了RNA-seq分析,并与有应答小鼠和未处理小鼠进行了比较。通路富集分析显示,相对于未处理小鼠和应答小鼠,无应答小鼠肿瘤中的Wnt通路上调,免疫反应下调(图 7C)。与此数据一致,对肿瘤提取物的Western印迹分析表明,在所有处理组中,相对于应答小鼠,无应答肿瘤中活性 β-catenin的表达增加,H3K18la恢复(图7D)。对PTEN/p53缺陷肿瘤来源的AC1/SC1细胞进行的体外WB分析表明,相对于24小时处理,在AD条件下,copanlisib + aPD-1处理72小时后,AC1/SC1细胞中活性β-catenin的表达量会同时增加。Wnt/β-catenin 信号传导的这种反馈激活伴随着乳酸生成量在72小时内的增加,而在体外进行急性 copanlisb/AD/aPD-1联合处理后,乳酸生成量最初会减少(图7E-F)。重要的是,在AD/copanlisib/aPD-1联合处理后 72 小时收集的体外CM对BMDM的处理显示,与24小时CM处理相比,吞噬细胞活性降低,H3K18la增加。总之,这些数据表明,Wnt-β-catenin通路的激活以及通过乳酸分泌和组蛋白乳酸化恢复肿瘤细胞/TAM串扰驱动了对ADT/PI3Ki/aPD-1联合疗法的耐药性(图7G)。
四 小 结
作者研究结果表明,在PTEN/p53缺陷的小鼠PC中,Wnt/β-catenin通路的激活通过恢复肿瘤细胞的乳酸生成和由此产生的TAM抑制来驱动对PI3Ki处理的耐药性。先前的研究表明,APC突变型癌症通过上调Wnt途径从肿瘤细胞产生更多的乳酸。此外,PI3Ki处理可增加Wnt-ligands的转录和共受体LRP5/6的磷酸化,从而拆除β-catenin降解复合物,导致Wnt/β-catentin途径激活介导的结直肠癌耐药。这些发现突出了致癌信号通路之间的相互作用,以保持Warburg效应,当精准药物处理靶向PI3K通路时,Warburg效应有助于免疫代谢耐药性的发展。总之,作者的研究表明,添加Wnt/β-catenin通路抑制剂,与ADT/PI3Ki/PD-1阻断疗法相结合,可以潜在地克服耐药性。
原文链接:https://doi.org/10.1158/1078-0432.CCR-22-3350