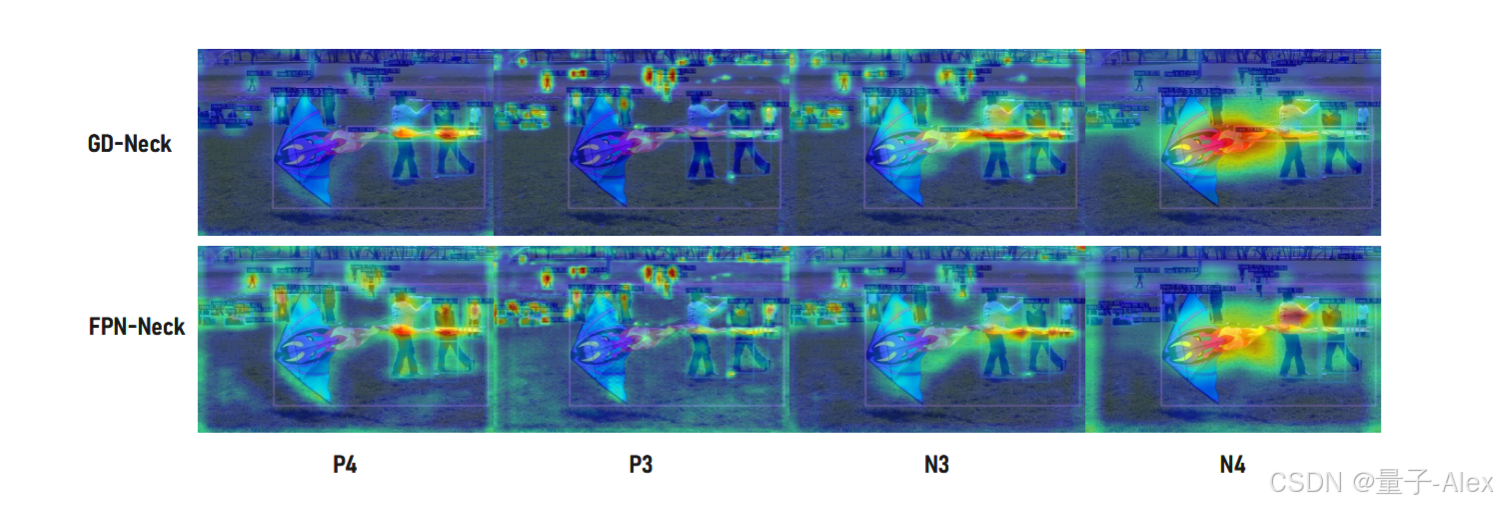
FSM-3:串行序列
- 1 Serial receiver
FSM使用总结:
- 所有涉及输出的driver原则上用cur_sta;若是使用nxt_sta的相当于是提前一拍知道结果,所以对于输出必须要使用clocked reg,这样才能和cur_sta对应起来;
- 描述声明状态按照cur_sta的为准,这样输出才能按照cur_sta的为准;
1 Serial receiver


module top_module(input clk,input in,input reset, // Synchronous resetoutput done
); parameter S0 = 5'b0_0000; //Startparameter S1 = 5'b0_0001; //Dataparameter S2 = 5'b0_0010; //Stopparameter S3 = 5'b0_0100; //OKparameter S4 = 5'b0_1000; //Errorparameter S5 = 5'b1_0000; //wait_finishreg[5 -1:0] cur_sta;reg[5 -1:0] nxt_sta;//==State transitionalways @(*) begincase(cur_sta)S0: nxt_sta = (in==1'b0) ? S1: S0;S1: nxt_sta = (cnt==7) ? S2 : S1;S2: nxt_sta = (in==1'b1) ? S3 : S4;S3: nxt_sta = (in==1'b1) ? S0 : S1;S4: nxt_sta = (in==1'b1) ? S0 : S5;S5: nxt_sta = (in==1'b1) ? S0 : S5;default : nxt_sta = S0;endcaseend//==State Flop-Flopalways @(posedge clk) beginif(reset) begincur_sta <= S0;end else begincur_sta <= nxt_sta; endend//==State Outputreg[8 -1:0] cnt;always @(posedge clk) beginif(reset) begincnt <= 0;end else begincase(cur_sta) S0: cnt <= 0;S1: cnt <= cnt + 1;S2: cnt <= 0;default: cnt <= 0;endcaseendendassign done = (cur_sta==S3); endmodule题目:

module top_module(input clk,input in,input reset, // Synchronous resetoutput [7:0] out_byte,output done
); //// Use FSM from Fsm_serialparameter S0 = 5'b0_0000; //Startparameter S1 = 5'b0_0001; //Dataparameter S2 = 5'b0_0010; //Stopparameter S3 = 5'b0_0100; //OKparameter S4 = 5'b0_1000; //Errorparameter S5 = 5'b1_0000; //wait_finishreg[5 -1:0] cur_sta;reg[5 -1:0] nxt_sta;//==State transitionalways @(*) begincase(cur_sta)S0: nxt_sta = (in==1'b0) ? S1: S0;S1: nxt_sta = (cnt==7) ? S2 : S1;S2: nxt_sta = (in==1'b1) ? S3 : S4;S3: nxt_sta = (in==1'b1) ? S0 : S1;S4: nxt_sta = (in==1'b1) ? S0 : S5;S5: nxt_sta = (in==1'b1) ? S0 : S5;default : nxt_sta = S0;endcaseend//==State Flop-Flopalways @(posedge clk) beginif(reset) begincur_sta <= S0;end else begincur_sta <= nxt_sta; endend//==State Outputreg[8 -1:0] cnt;always @(posedge clk) beginif(reset) begincnt <= 0;end else begincase(cur_sta) S0: cnt <= 0;S1: cnt <= cnt + 1;S2: cnt <= 0;default: cnt <= 0;endcaseendendassign done = (cur_sta==S3); // New: Datapath to latch input bits.reg[8 -1:0] data;always @(posedge clk) beginif(reset) begindata <= 0;end else begincase(cur_sta)S1: data[cnt] <= in;S2: data <= data;default: data <= data;endcaseendendassign out_byte = (done==1) ? data : 0;endmodule