小罗碎碎念
本期推文的主题:人工智能在空间转录组学领域的最新进展
提到空间转录组学就不可避免会与单细胞测序、免疫治疗以及肿瘤微环境扯上关系,所以这也是当下的热点之一。
我个人认为这一部分门槛相对于影像组学和病理组学较高,需要具备比较系统的临床知识,才能hold住这个课题。我之前一直在下意识的避开这一部分,我觉得不好,所以准备了这期推文。
我是罗小罗同学,明天见!!

一、晚期透明细胞肾细胞癌(aRCC)患者接受免疫检查点阻断(ICB)治疗的多组学研究

文献概述
这篇文章是一项关于晚期透明细胞肾细胞癌(aRCC)患者接受免疫检查点阻断(ICB)治疗的多组学研究。
研究团队通过综合分析肿瘤的多组学数据,开发了一种新的肿瘤转录组特征,即低人类白细胞抗原(HLA)抗原肽结合特异性(HLAprLOW)的肿瘤特征,这种特征能够预测患者对ICB治疗的临床反应。
研究包括了对超过1000名患者的肿瘤转录组数据的分析,并通过单细胞和空间分辨率的验证,揭示了患者特异性的炎症性肿瘤相关巨噬细胞和(前)耗竭的CD8+ T细胞之间的相互作用。这些细胞特征通过一个交叉组学生物信息学机器学习管道得到了进一步的分析,并与ICB治疗后的积极结果相关联。
此外,研究还使用了RENCA肿瘤小鼠模型进行实验,发现CD40激动剂与PD1阻断相结合可以增强肿瘤相关巨噬细胞和CD8+ T细胞的炎症反应,从而实现相对于其他测试方案的最大抗肿瘤效果。
研究结果表明,通过多组学和空间图谱可以更好地理解驱动ICB反应的免疫社区结构,并为aRCC患者的临床治疗提供了新的生物标志物和治疗策略。
重点关注
图1提供了对Leuven RWD队列中晚期透明细胞肾细胞癌(aRCC)患者进行免疫检查点阻断(ICB)治疗研究的概览。
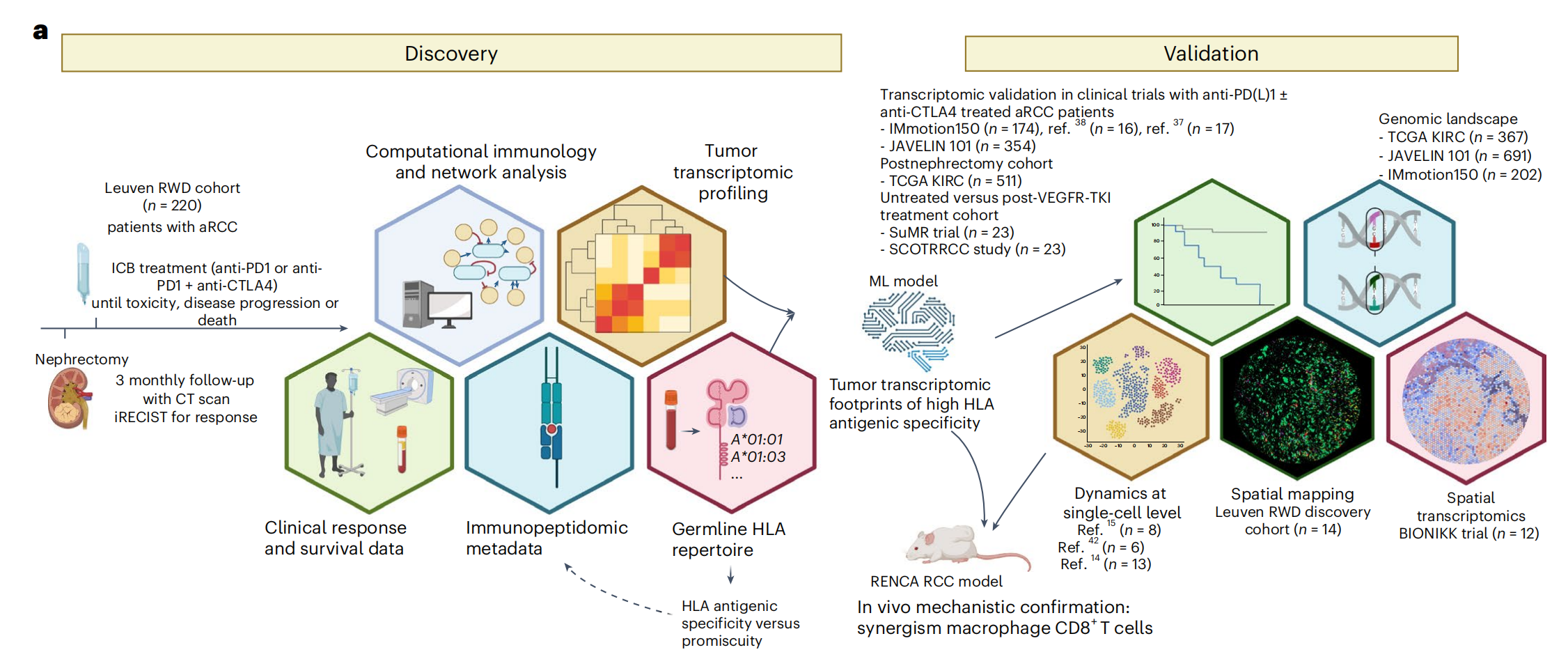
a. 研究设计概览:描述了研究的发现阶段,首先是在Leuven RWD队列中进行,之后使用机器学习(ML)模型开发了一个生物标志物,然后这个标志物在外部(批量)数据集以及单细胞和空间层面上进行了验证,并且从基因组学角度进行了表征。最后,通过小鼠RCC/RENCA模型提供了体内功能性验证。
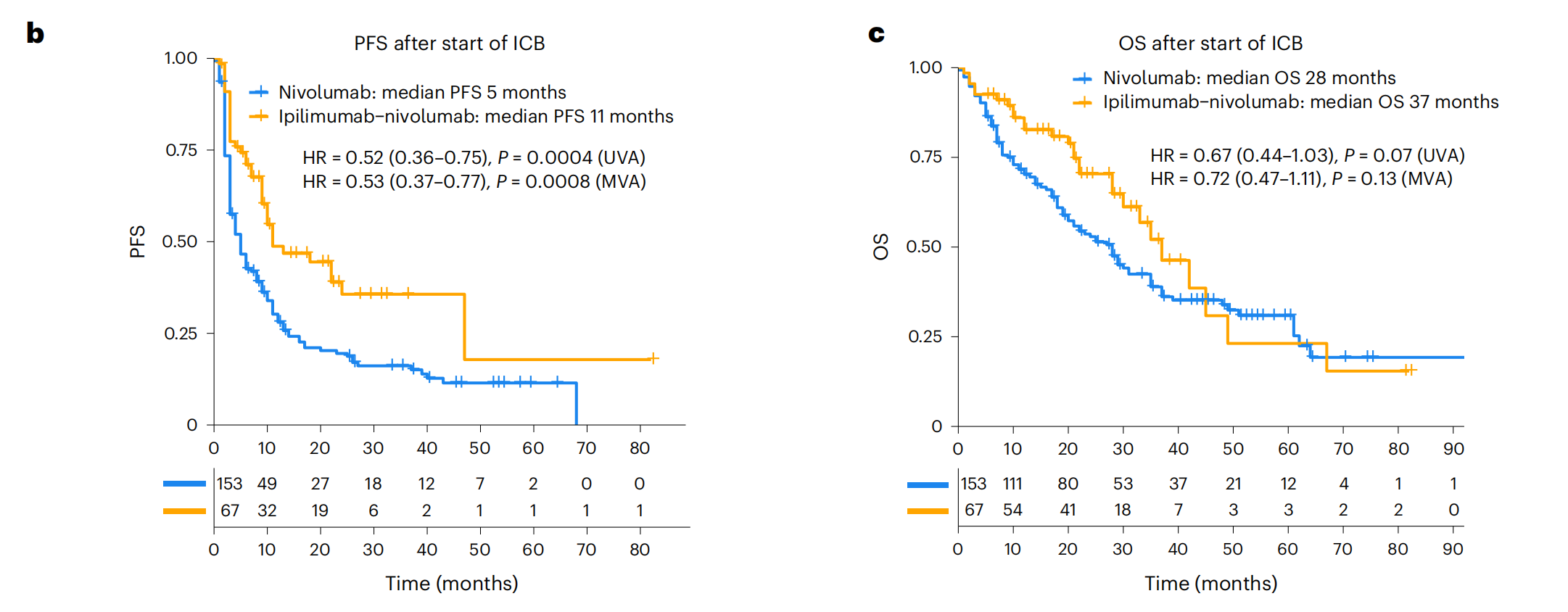
b. PFS的Kaplan-Meier曲线:展示了Leuven RWD队列中ICB治疗开始后无进展生存期(PFS)的Kaplan-Meier曲线,根据ICB治疗类型(ipilimumab-nivolumab与nivolumab)进行了分层。使用Cox比例风险回归模型计算了风险比(HR)和置信区间(CI),比较了接受ipilimumab-nivolumab治疗与仅接受nivolumab治疗的患者。
c. OS的Kaplan-Meier曲线:与b部分类似,但展示的是总生存期(OS)的Kaplan-Meier曲线,同样根据ICB治疗类型进行了分层,并计算了HR和CI。
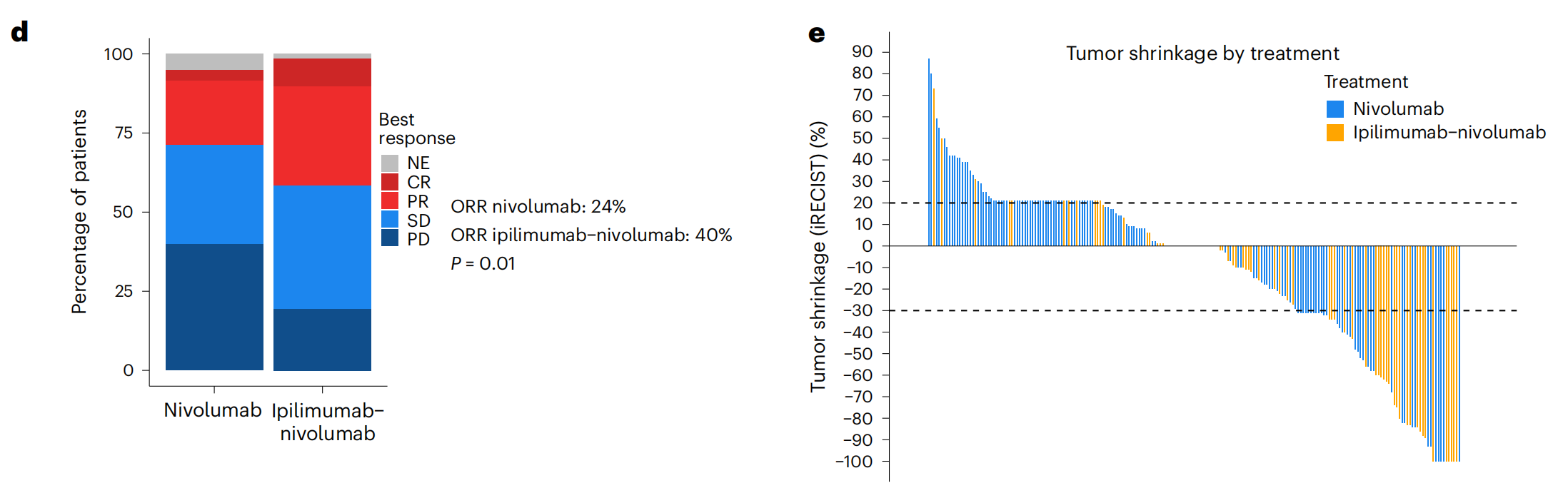
d. 最佳反应的堆叠条形图:通过iRECIST评估标准,展示了ICB治疗的最佳反应,包括完全缓解(CR)、部分缓解(PR)、疾病稳定(SD)、疾病进展(PD)和不可评估(NE)。使用Fisher精确检验计算了两组之间的P值。
e. 瀑布图:展示了每位患者通过iRECIST评估的最大肿瘤缩小情况。CT指的是计算机断层扫描,用于评估肿瘤大小的变化。
整体来看,图1提供了对ICB治疗反应的全面评估,包括生存数据、治疗反应的统计分析以及不同治疗组之间的比较。这些数据对于理解ICB治疗在aRCC患者中的临床效果至关重要。
二、BIDCell:专为亚细胞空间转录组数据的细胞分割而设计的新型自监督深度学习框架

文献概述
这篇文章介绍了一种名为BIDCell的新型自监督深度学习框架,它专为亚细胞空间转录组数据的细胞分割而设计。
BIDCell利用生物学信息丰富的损失函数,学习空间分辨基因表达与细胞形态之间的关系。该框架结合了单细胞转录组数据和细胞形态信息,通过全面的评估框架,证明了BIDCell在多种组织类型和技术平台上,根据不同的细胞分割性能指标,优于其他最先进的方法。
BIDCell的关键创新在于其生物学信息损失函数的多组分协同作用,以及明确地整合单细胞测序数据中的先验知识,以估计不同细胞形态。这些损失函数和使用现有scRNA-seq数据的结合提高了性能,并且BIDCell能够跨不同SST平台通用。此外,作者还创建了一个全面的细胞分割性能评估框架CellSPA,涵盖五个互补类别的标准,旨在促进新分割方法在新型生物技术数据中的采用。
研究结果表明,BIDCell在改善细胞形态表示方面表现优异,能够生成更多样化的细胞形态特征,并克服了现有方法的局限性,这些现有方法主要依赖于SST图像强度值进行细胞分割。BIDCell还通过利用核内的表达模式引导细胞体像素的识别,并通过消融研究展示了不同损失函数的协同效应。
文章还展示了BIDCell在多个SST平台上的复制性和泛化能力,并通过案例研究进一步评估了BIDCell在准确分割紧密堆积的细胞方面的性能。最后,作者讨论了BIDCell在亚细胞空间分辨转录组数据中的细胞分割方法的潜力,以及CellSPA评估框架在促进可重复科学和透明度方面的重要性。
重点关注
Fig. 1展示了BIDCell框架及其用于训练的损失函数的示意图。
这个框架是一个自监督的深度学习模型,专门设计用于细胞分割任务,尤其是在处理空间转录组数据时。
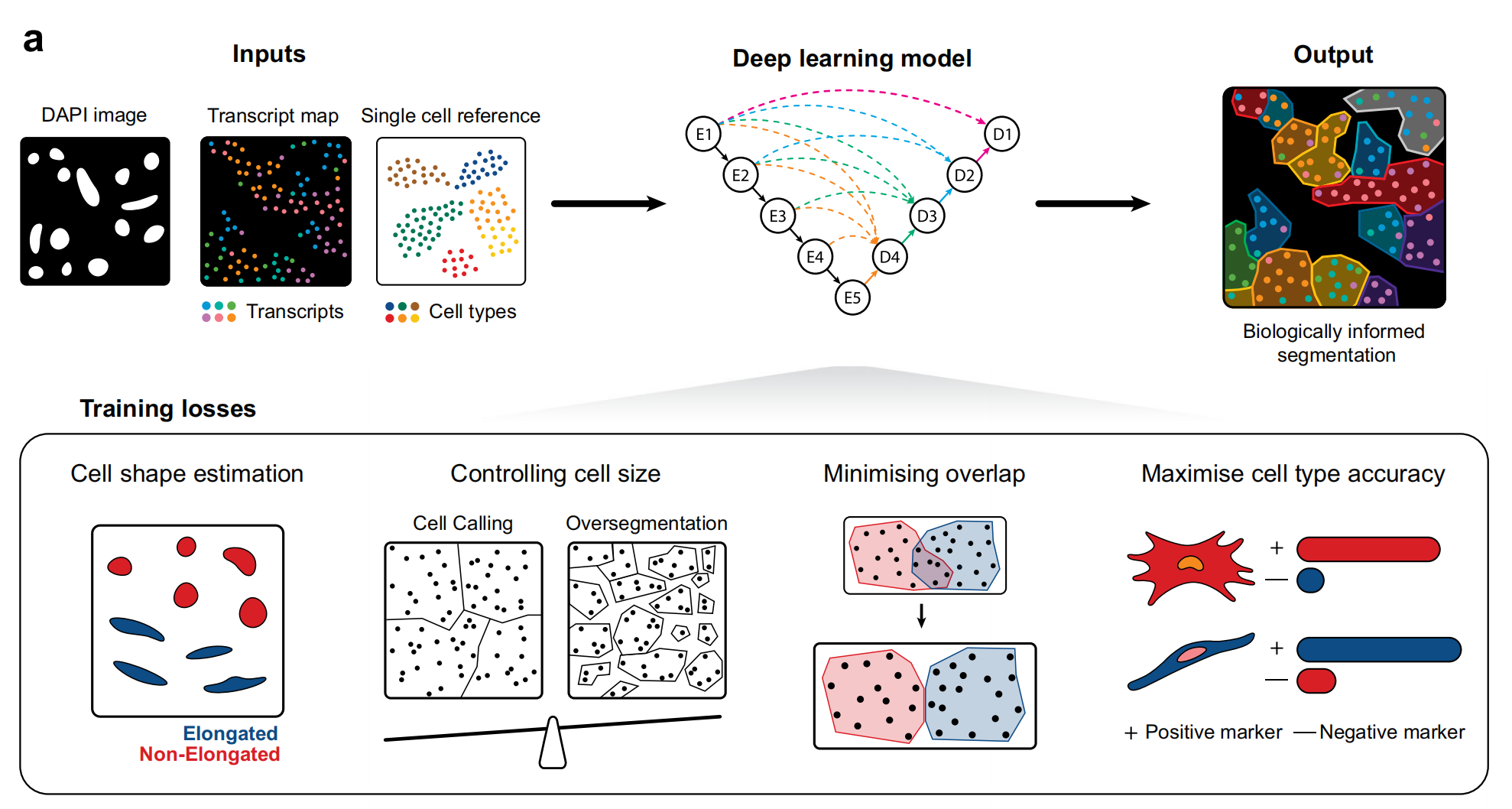
a部分 描述了BIDCell框架的结构:
- 框架中包含编码层(E1到E5)和解码层(D1到D4),这些层负责提取特征并重建细胞分割图。
- 每一层解码层都与多个编码层相连,以恢复图像的细节,这种连接通过不同颜色的箭头表示,例如D3层用绿色箭头表示。
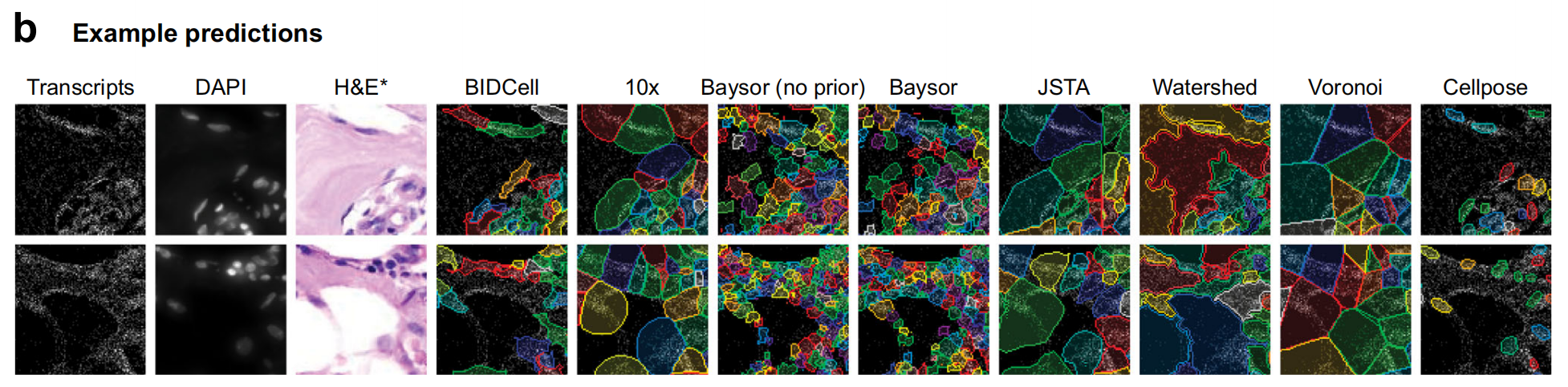
b部分 对比了BIDCell与其他细胞分割方法在公共Xenium-BreastCancer1数据集上的预测结果:
- BIDCell能够更好地捕捉与输入图像相对应的细胞形态,包括更多样化的细胞形状,例如长条形状的细胞。
- 该图还展示了BIDCell生成的分割结果与其他方法(如10x Genomics、Baysor、Cellpose等)的结果的对比。
- H&E图像(一种常用的组织染色技术)仅用于说明目的,并未作为任何方法的输入。
总体而言,Fig. 1强调了BIDCell在处理复杂细胞形态时的优势,尤其是在识别和分割具有不同形状和大小的细胞方面。通过自监督学习,BIDCell能够利用生物学信息和空间基因表达数据来提高分割的准确性和生物学相关性。
三、数字病理学和空间组学技术在脂肪肝病的临床应用和潜力

文献概述
这篇文章主要探讨了数字病理学(Digital Pathology, DP)和空间组学(Spatial Omics)技术在脂肪肝病(Steatohepatitis)的临床应用和发现潜力。
文章首先介绍了脂肪肝病的背景,包括其定义、病因和组织学表现。接着,讨论了数字病理学如何通过将传统的病理切片数字化,结合人工智能(AI)算法来提高组织学解读的准确性和速度。
此外,文章还探讨了空间生物学的新领域,该领域允许研究者在肝组织切片上特定感兴趣区域(Region of Interest, ROI)内映射基因和蛋白质的表达,从而理解疾病在组织内的异质性和分子变化与特定组织形态之间的关系。
文章强调了多模态整合的重要性,即将传统的临床数据与AI增强的DP和空间生物学结合起来,以提高脂肪肝病诊断的精确度和组织学终点评估。最后,文章讨论了当前知识的差距、这些工具和技术的局限性与前提,并展望了未来的研究方向。
关键点包括:
- 尽管肝活检是诊断脂肪肝病的金标准,但MASLD/MASH与酒精相关肝病之间没有特异的组织学特征。
- 数字病理学的发展,以及机器学习(ML)算法的应用,显著提高了从诊断角度对脂肪肝病组织学解读的准确性和速度,并大大增加了肝活检的发现潜力。
- 空间组学技术的快速发展,为基于对基因、蛋白质和代谢物表达的广泛分析,与肝组织形态相关的空间生物学新领域奠定了基础。
- 空间生物学目前被用作发现工具,用于解码肝脏免疫微环境、细胞异质性和细胞间信号通路,以识别脂肪肝病中的新型病理介质、疾病生物标志物和治疗靶点。
- 传统临床数据与AI增强的DP和空间生物学的多模态整合,预计将提高脂肪肝病诊断的精确度和组织学终点评估。
文章由Mayo Clinic的研究人员撰写,并得到了美国国立卫生研究院(NIH)的支持。作者声明没有利益冲突。
重点关注
图2展示了两种评估和量化脂肪肝病组织病理特征和纤维化的方法的示意图。
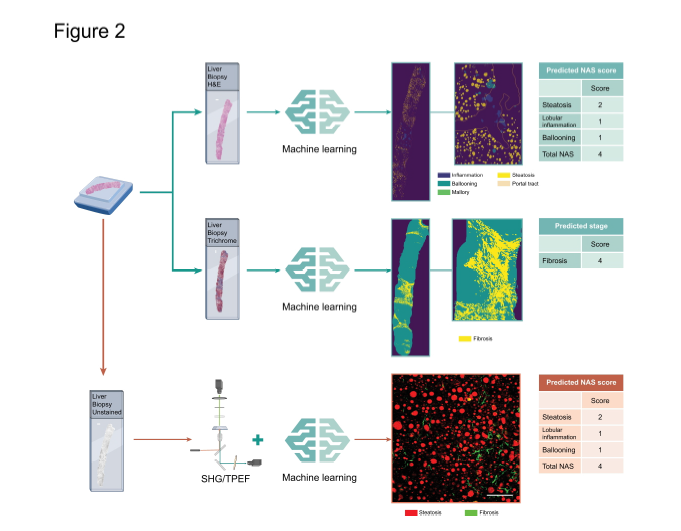
以下是对这两种方法的分析:
-
基于机器学习算法的组织学特征量化:
- 该方法首先使用机器学习(ML)算法对苏木精-伊红(H&E)染色的玻片进行分析,量化脂肪肝病的三个关键组织学特征:脂肪变性(steatosis)、小叶性炎症(lobular inflammation)和气球样变(ballooning)。
- 这种基于ML的分析可以集成到日常的工作流程中,允许病理学家验证ML算法的预测结果。
-
基于非线性光学显微镜的非破坏性量化:
- 第二种方法利用单张未染色的玻片,通过非破坏性方式进行分析,使用二次谐波生成(SHG)和双光子激发荧光(TPEF)技术来量化脂肪肝病的相同组织学特征,包括脂肪变性、炎症、气球样变和纤维化。
- 这种方法的优势在于它不需要对组织进行染色,因此不会破坏样本,允许进行额外的分析或保留原始组织状态。
两种方法的共同点在于它们都旨在提高组织病理学评估的准确性和效率。第一种方法侧重于利用ML算法处理常规染色的玻片,而第二种方法则侧重于使用先进的光学技术对未染色的组织样本进行分析。两种方法都为病理学家提供了工具,以更客观、更精确地评估脂肪肝病的组织学特征,从而可能改善诊断和治疗决策。
四、stKeep:分析空间分辨的转录组学数据,精确解析肿瘤微环境的新型异构图学习方法

文献概述
这篇文章介绍了一种名为stKeep的新型异构图学习方法,用于分析空间分辨的转录组学数据(Spatially Resolved Transcriptomics, SRT),以精确解析肿瘤微环境(Tumor Microenvironment, TME)。
stKeep通过整合多模态数据和基因-基因相互作用,能够识别TME中的细胞模块、基因模块和细胞间通信(Cell-Cell Communication, CCC)。该方法利用异构图学习来捕捉细胞、基因和组织区域之间复杂的关系,从而在低维空间中检测更多的细胞状态和细胞状态特异性的基因-基因关系。此外,stKeep还使用对比学习来推断每个细胞的CCC模式,确保学习到的CCC模式在不同的细胞状态之间具有可比性。
研究者们使用stKeep分析了多种癌症样本,包括乳腺癌、肺癌、结直肠癌和肝转移瘤,发现它在解析TME方面优于其他工具。stKeep能够识别关键的转录因子、配体和受体,这些与疾病进展相关,并且通过独立临床数据的功能和生存分析进一步验证了其临床预后和免疫治疗应用的潜力。
文章还讨论了stKeep在处理SRT数据时的挑战,如低通量、灵敏度以及数据的稀疏性和噪声问题。通过与其他方法的比较,证明了stKeep在分析人类背外侧前额叶皮层(DLPFC)数据集时的性能。此外,stKeep还有助于识别具有生物学意义的细胞模块和基因模块,这对于理解肿瘤生态系统的复杂性至关重要。
最后,文章讨论了stKeep的潜在应用,包括在不同癌症类型中解析异质性TME,识别与疾病进展和远处转移相关的细胞状态和机制,并可能揭示抗癌治疗的新靶点。作者还指出了stKeep的局限性,并提出了未来改进的方向,包括开发更复杂的算法来推断更多的基因-基因关系及其方向,以及自动化分割肿瘤区域的计算模型。
重点关注
Fig. 1 提供了 stKeep 模型的概述,该模型是一个用于分析肿瘤生态系统的异构图学习方法。
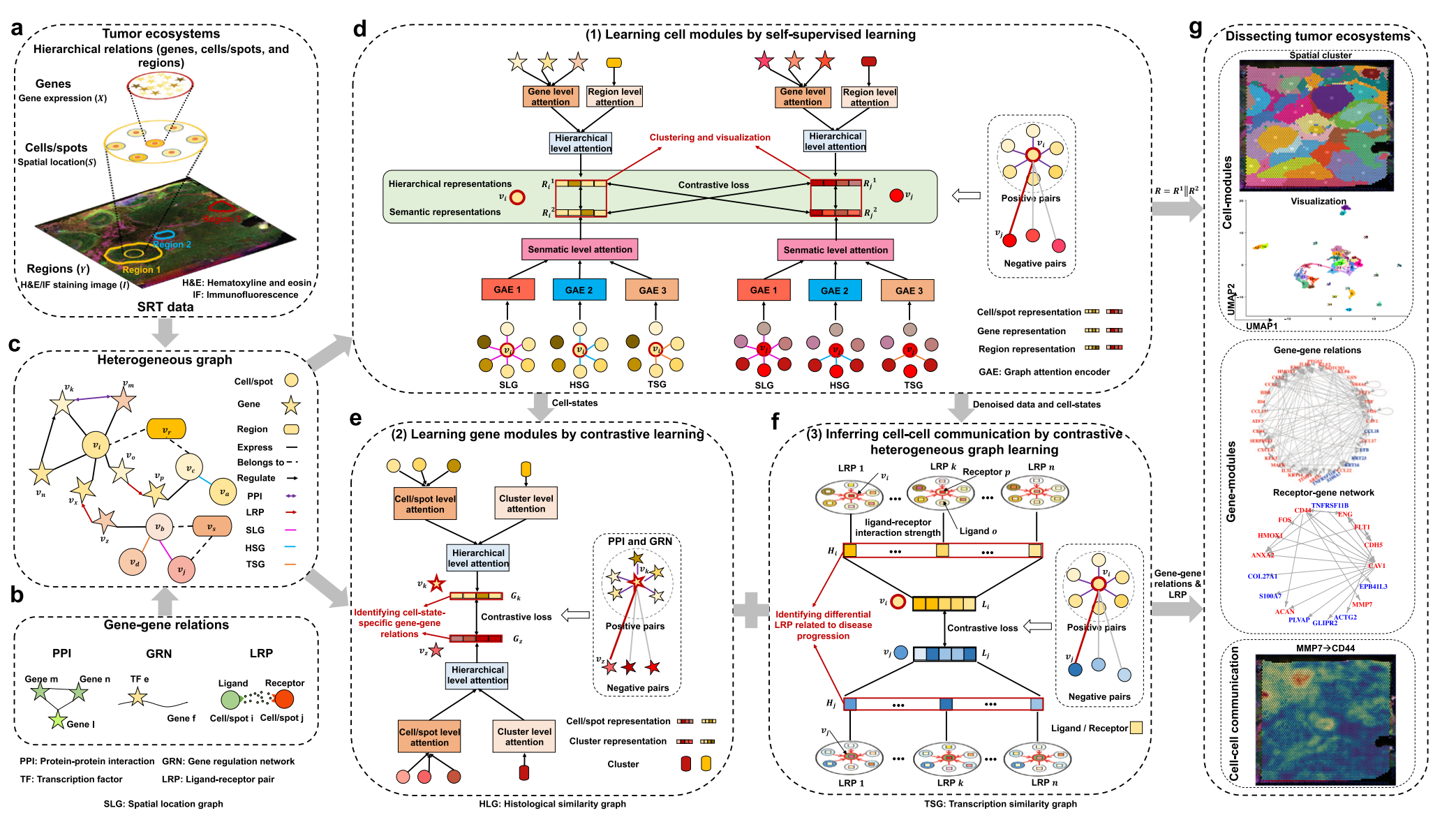
下面是对图示的分析:
a–c. stKeep 模型接收四层 SRT 数据作为输入:
- 组织学图像 (I):提供了细胞和组织的空间结构信息。
- 空间位置 (S):记录了细胞或斑点在组织中的具体位置。
- 基因表达 (X):包含了细胞或斑点中基因表达的数据。
- 组织学区域 (Y):定义了不同的组织区域,有助于区分不同的细胞状态或类型。
此外,模型还考虑了基因-基因之间的相互作用,如蛋白质-蛋白质相互作用 (PPI)、基因调控网络 (GRN) 和配体-受体对 (LRP)。
d. 细胞模块:
- 以细胞/斑点为中心的异构图 (HG) 用于捕捉局部层次表示 (Ri1),通过注意力机制聚合来自基因和区域的特征。
- 利用多种语义图,包括空间位置图 (SLG)、组织学相似性图 (HSG) 和转录相似性图 (TSG) 来学习全局语义表示 (Ri2)。
- 通过自监督学习将局部层次和全局语义表示整合起来。
e. 基因模块:
- 以基因为中心的异构图用于学习低维表示,通过注意力机制结合来自细胞/斑点和簇的特征。
- 使用对比学习确保在表示空间中相关联的基因对彼此邻近。
f. 细胞间通信 (CCC) 模块:
- 利用基于注意力的异构图来推断每个细胞/斑点的配体-受体相互作用强度 (Hi)。
- 通过聚合中心细胞/斑点周围邻居的配体信息来推断 CCC,同时确保学习到的 CCC 模式能够在不同的细胞状态中体现出差异。
g. 统一框架:
- 结合了细胞模块、基因模块和 CCC 模块的统一框架,可以用于:
- 检测和可视化空间簇。
- 识别细胞状态特异性的基因模块和受体-基因交互网络。
- 推断细胞间的通信强度。
总体而言,Fig. 1 展示了 stKeep 模型如何整合多模态数据和多种生物分子相互作用,通过构建异构图来解析肿瘤微环境中的复杂相互作用,进而识别细胞状态、基因模块和细胞间通信模式。
五、使用机器学习集成方法推进计算免疫学的研究

文献概述
这篇文章是关于如何使用机器学习集成方法来推进计算免疫学的研究。
文章首先指出传统的免疫学研究依赖于蛋白质组学来评估单个免疫细胞,但随着单细胞RNA测序技术的发展,计算免疫学家在分析这些数据集时,已经超越了传统的蛋白质标记识别,能够更详细地观察细胞表型及其功能角色。
文章强调了最近技术进步允许在单细胞水平上同时测量多种细胞组分,包括转录组、蛋白质组、染色质、表观遗传修饰和代谢物,这导致了复杂多尺度数据集的生成。现代机器学习技术可以整合多种“组学”数据,无需对每种模式进行广泛的独立建模。
文章重点介绍了应用于免疫学研究的ML集成方法的最新进展,并强调了这些方法在创建多尺度数据集合的统一表示中的重要性,特别是对于单细胞和空间分析技术。最后,文章讨论了这些全面方法的挑战,并展望了它们在发展多尺度研究的共同坐标框架中的作用,从而加速研究并促进计算免疫学领域的发现。
背景部分讨论了免疫系统是一个复杂的细胞网络,共同协调对未识别干扰的防御反应。文章提到免疫细胞设计上不局限于特定位置,它们已经适应在各种微环境中运作,并在健康和疾病条件下发挥作用,使得免疫学成为一个不断变化的研究领域。研究揭示了免疫系统在诸如神经疾病、糖尿病和癌症等复杂疾病中的重要作用。
文章还讨论了单细胞测量在免疫学中并不新鲜,但单细胞测序技术的出现极大地推动了该领域的发展。新技术允许对多种分子读数进行测量,包括转录组、表面和细胞内蛋白质组、染色质、表观遗传修饰、免疫库和代谢物。文章提到了一系列空间蛋白质组学技术的发展,这些技术结合了显微镜成像和基因表达,同时保留了空间位置信息。
在数据整合方面,文章提到了线性模型和深度学习方法,包括自动编码器、变分自动编码器、图神经网络和生成对抗网络等。这些方法被用于数据的降维、去噪和细胞状态的识别。
最后,文章讨论了将单细胞数据与空间分析数据整合的方法,以及如何预测特征表达的空间分布和空间数据的细胞类型反褶。文章还强调了创建共同参考框架的重要性,以及数据基础设施和多学科团队在处理多模态数据时的需求。
文章的结论是,使用多模态技术,免疫学家已经收集了大量的多尺度测量数据。整合和解释这些数据的ML方法对于理解健康和疾病中免疫系统的微妙调节机制至关重要。作者呼吁开发者确保方法的可解释性、跨多种条件的基准测试的可用性以及用例的文档化。文章还展望了未来,认为计算免疫学家将通过利用整合方法推进科学进步,发展个性化的免疫学干预措施,包括疫苗设计、免疫疗法和治疗计划,这些措施将精确调整以适应个体的免疫系统概况。
重点关注
Fig. 2 A和Fig. 2 B提供了多模态数据整合方法和数据存储结构的示意图。
Fig. 2 A
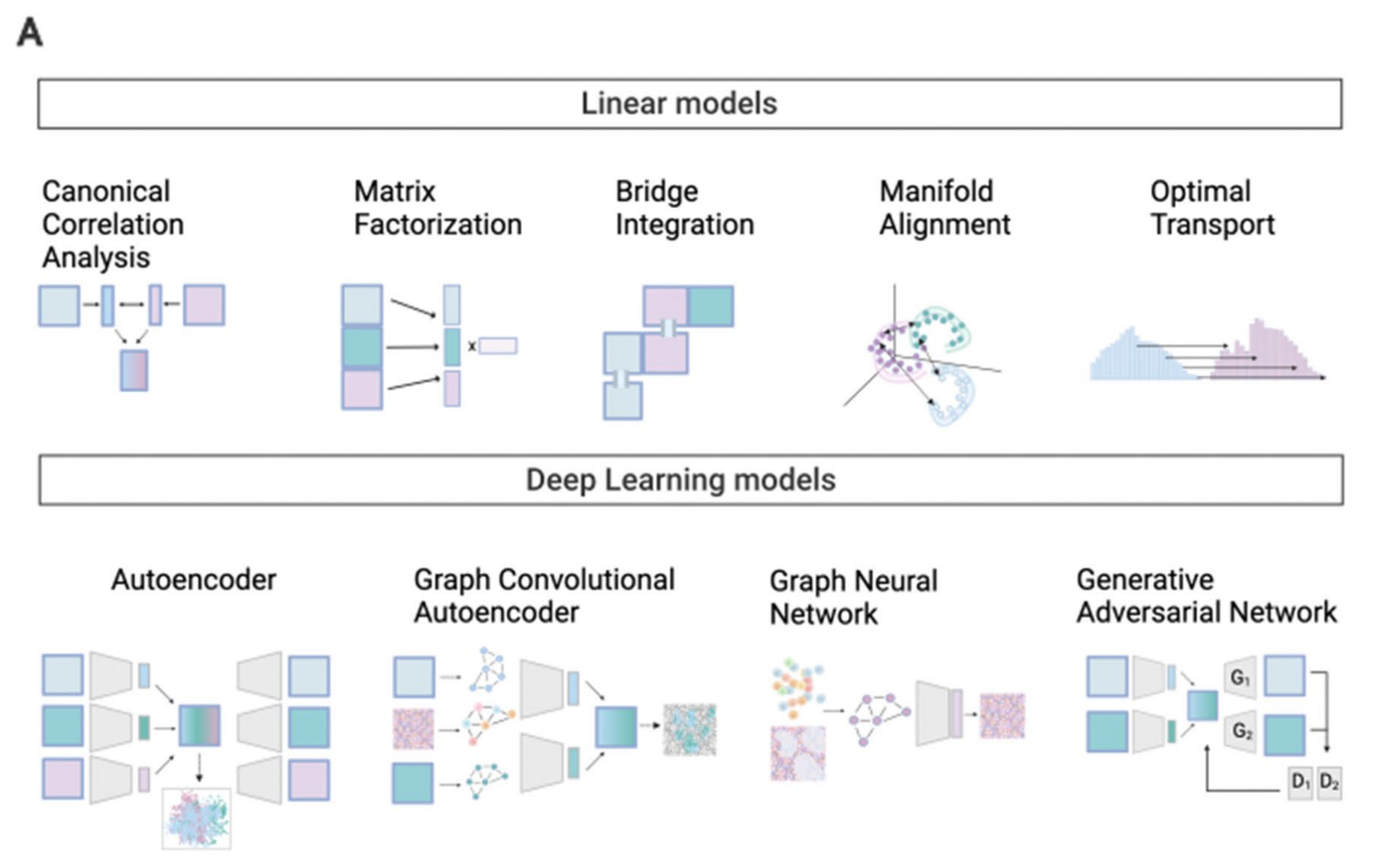
展示了代表性的多模态数据整合方法的示意图。这可能包括不同的算法和技术,用于处理和分析来自不同来源和具有不同特性的数据。整合方法的目的是将这些数据集融合在一起,以便于能够从整体上理解复杂的生物系统。例如,可能包括线性模型、深度学习架构、生成对抗网络(GANs)、自动编码器(AEs)等,这些方法可以帮助识别数据中的模式和关联,从而为生物医学研究提供洞察。
Fig. 2 B
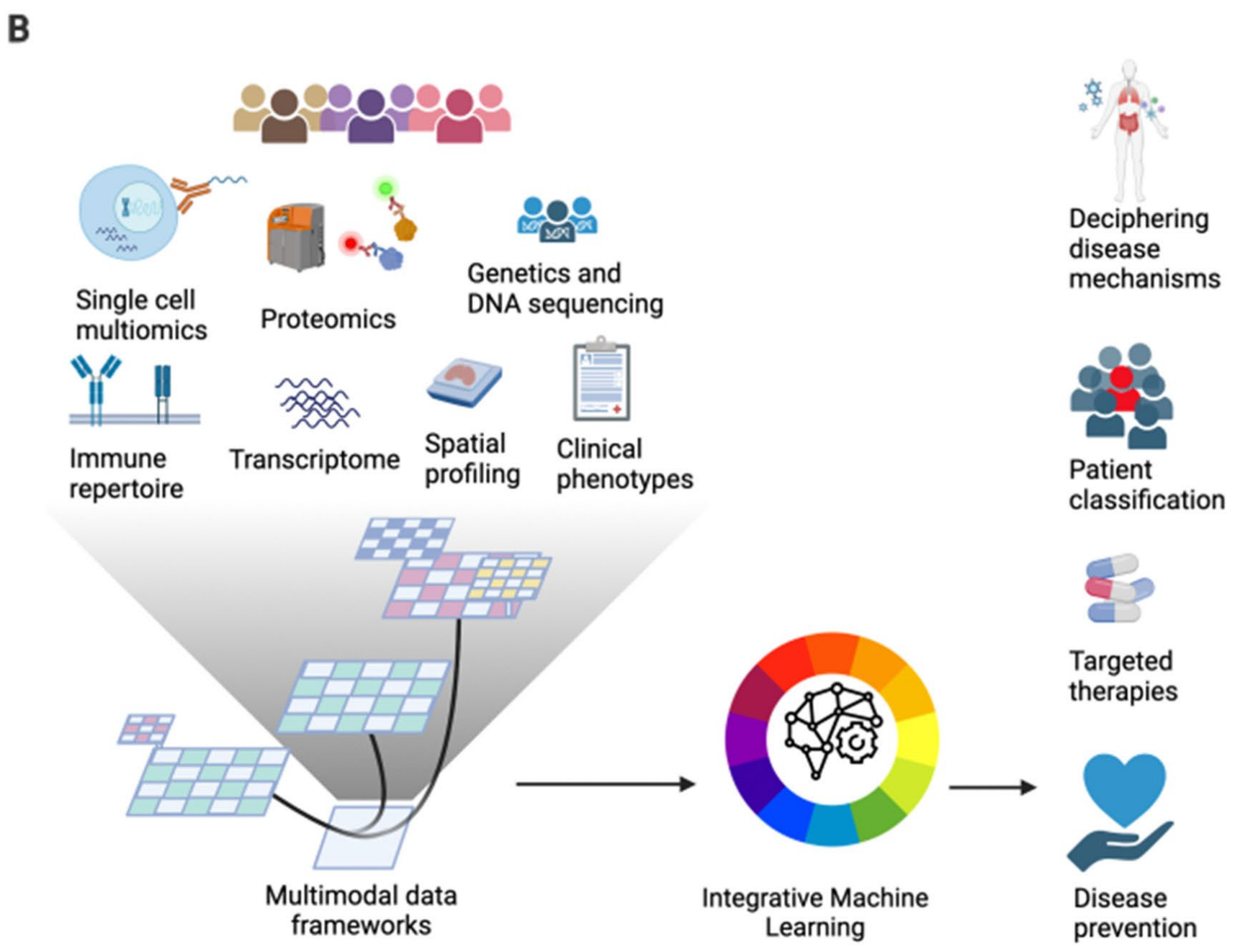
描述了多模态数据集具有不同的数据格式,并且可以存储在专门的数据结构中,这些数据结构允许高效地独立或联合访问和处理每个层面的数据。这意味着数据可以以一种方式组织,使得研究人员可以针对他们的特定分析需求,快速检索和操作数据。例如,单细胞技术可能产生稀疏矩阵,其行和列分别代表细胞和特征(基因、蛋白质、染色质区域),而空间分析技术可能提供亚细胞或细胞聚合的分子分析,以及组织的图像。
这些数据基础设施构成了机器学习整合方法的核心,这些方法反过来又提供了多样化的生物医学见解。换句话说,通过有效地存储和组织数据,可以应用机器学习技术来整合和分析数据,从而揭示生物学过程的新见解。整合方法不仅有助于理解单个数据集,而且有助于在不同数据类型之间建立联系,这对于全面理解复杂疾病和生物系统至关重要。
总的来说,这两个图表强调了数据整合在现代生物医学研究中的重要性,以及如何通过先进的数据处理和机器学习技术来实现这一目标。
六、关于非小细胞肺癌的单细胞和空间转录组学分析研究

文献概述
这篇文章是一项关于非小细胞肺癌(non-small cell lung cancer, NSCLC)的单细胞和空间转录组学分析研究。
研究团队由Marco De Zuani、Haoliang Xue等人组成,他们对25位未经治疗的肺腺癌和鳞状细胞癌患者的约90万个细胞进行了深入分析。研究发现肿瘤生态系统中存在多种免疫细胞类型,尤其是髓系细胞,它们在疾病进展中扮演着重要角色。
研究揭示了抗炎性巨噬细胞与NK细胞/T细胞之间的负相关关系,以及肿瘤内NK细胞的细胞毒性降低。尽管肺腺癌和鳞状细胞癌在细胞类型组成上相似,但它们在多种免疫检查点抑制剂的共表达上存在显著差异。此外,研究还发现了肿瘤中巨噬细胞的转录“重编程”,使它们倾向于胆固醇外运,并采用类似胎儿的转录特征,促进铁的外流。
这项多组学资源提供了肿瘤相关巨噬细胞的高分辨率分子图谱,增进了我们对其在肿瘤微环境中角色的理解。研究结果不仅为理解肺腺癌和鳞状细胞癌的肿瘤微环境提供了新的视角,而且为开发新的治疗策略提供了可能的靶点,尤其是针对免疫检查点抑制剂和巨噬细胞的重编程过程。
文章还讨论了肿瘤微环境中的细胞间相互作用网络,以及不同NSCLC亚型(肺腺癌LUAD和肺鳞癌LUSC)之间在这些网络方面的差异。此外,研究还探讨了肿瘤巨噬细胞可能经历的“肿瘤胎儿重编程”,这可能与肿瘤生长和转移有关。
重点关注
图1展示了对非小细胞肺癌(NSCLC)异质性的单细胞转录组学分析。
以下是对图中各个部分的详细分析:
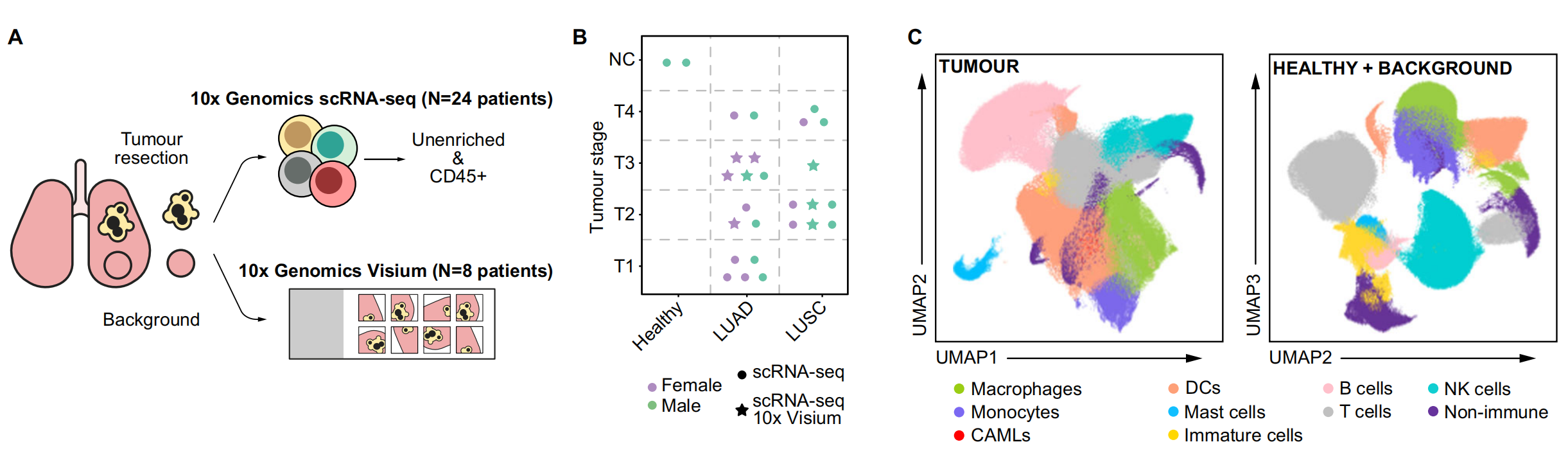
A. 研究概述:
- 研究使用了来自切除的肿瘤组织、邻近未受累组织(背景)和已故捐献者的健康肺组织的单细胞悬浮液。
- 这些细胞被富集为CD45+或CD235−,然后进行了单细胞RNA测序(scRNA-seq)。
- 同时,使用新鲜冷冻的肿瘤、背景和健康组织切片进行了10x Genomics Visium空间转录组学分析。
B. 队列概述:
- 使用符号代表个别患者和执行的分析。
C. UMAP投影:
- 展示了肿瘤和合并的背景+健康数据集的统一流形近似和投影(UMAP)。
D. 点图:
- 用于肿瘤样本中宽泛细胞类型注释的代表性基因点图。
E. 等高线图:
- 显示了在AT2细胞(44,399个细胞)、CAMLs(2520个细胞)和AIMɸ(16,120个细胞)中髓系(LYZ, CD68, MRC1)和上皮(EPCAM)基因的共表达。
- 基因表达经过了标准化、缩放和对数转换。
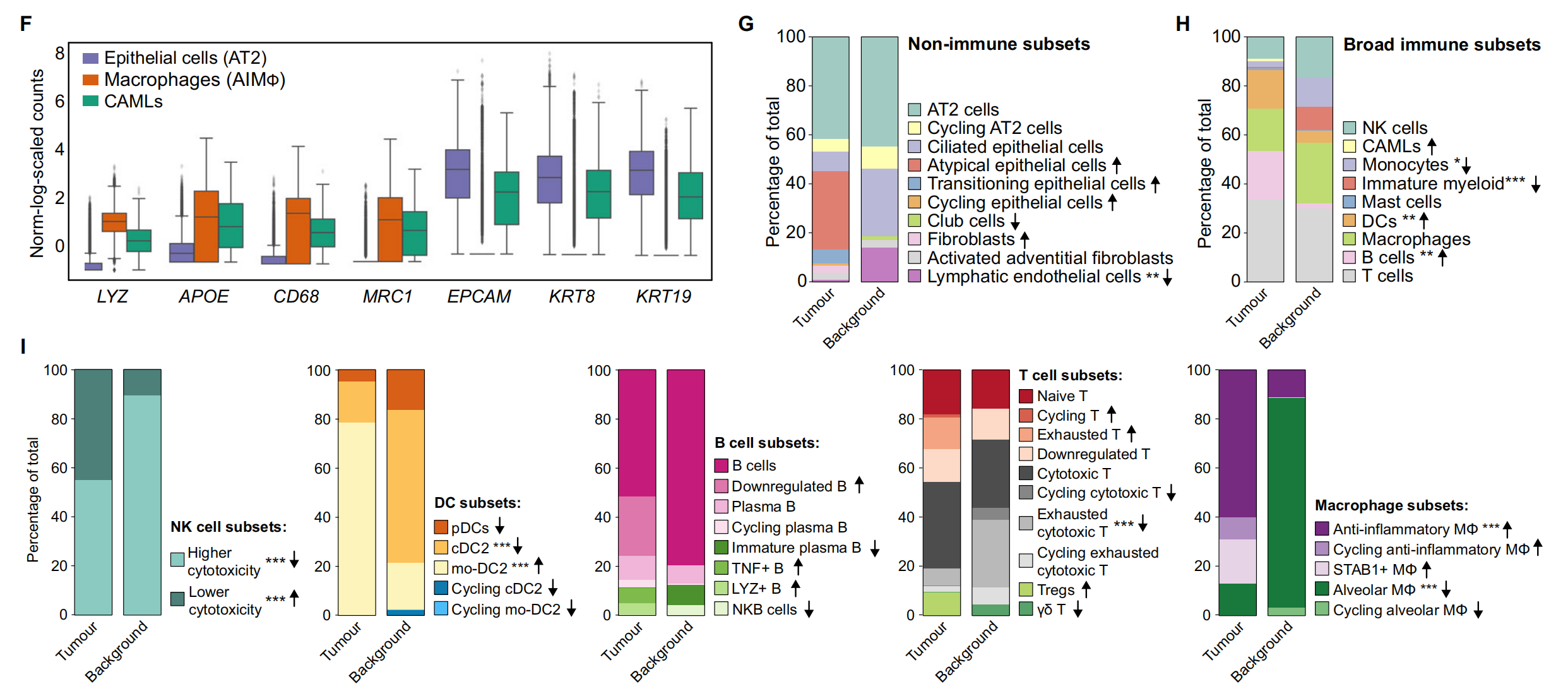
F. 箱线图:
- 展示了AT2细胞、CAMLs和AIMɸ中髓系(LYZ, APOE, CD68, MRC1)和上皮(EPCAM, KRT8, KRT19)基因的标准化、缩放和对数转换后的基因表达。
- 箱形图的箱线部分表示四分位数,须线表示1.5倍四分位距。
G. 非免疫细胞亚群的相对比例:
- 计算了肿瘤和背景组织中非免疫细胞亚群的相对比例,限定在CD235−富集的细胞中。
- 箭头表示肿瘤与背景相比的增加(↑)或减少(↓)。
- 使用双边Wilcoxon秩和检验和Bonferroni校正进行多重比较。
- **P < 0.01,没有星号的箭头表示该细胞类型仅在肿瘤或背景中发现。
H. 广泛免疫细胞的相对比例:
- 计算了肿瘤和背景组织中所有免疫细胞的相对比例,限定在CD235-富集的细胞中。
- 箭头和星号的使用同上,但P值的标准不同(*P < 0.05, **P < 0.01, ***P < 0.001)。
I. 肿瘤和背景中的NK、DC、B、T细胞和巨噬细胞亚群的相对比例:
- 计算了肿瘤和背景组织中NK细胞、树突细胞、B细胞、T细胞和巨噬细胞亚群的相对比例,限定在CD235-富集的细胞中。
- 统计方法和标记同上,但P值的标准为***P < 0.001。
整体来看,图1通过不同的统计图表和分析方法,展示了NSCLC肿瘤组织与邻近正常组织在细胞组成和基因表达上的差异,揭示了肿瘤微环境中的细胞异质性。