1写在前面
有小伙伴子留言问最近介绍的WGCNA共识网络的意义是什么,保守性吗!?🧐
与把雄性小鼠和雌性小鼠的数据merge在一起,一起构建网络、确定模块的方式有什么区别呢!?😗
其实区别还是挺大的,这种方式可以找到特异的模块,只属于雄性小鼠或雌性小鼠。🤓
不过生信分析本来就有其固有缺陷,最终还是需要实验来验证你的结果,所以分析方法的话仁者见仁,智者见智吧。🤒
OK,今天的教程是共识模块与特异模块相关联,也就是计算两者的overlap部分。😉
2用到的包
rm(list = ls())
library(tidyverse)
library(WGCNA)
3示例数据
我们这个时候要把前面清洗好,构建好的网络数据拿出来吧。😗
还要用到之前单独的雌性小鼠数据。🙂
load("./Consensus-dataInput.RData")
load("./Consensus-NetworkConstruction-auto.RData")
load("./FemaleLiver-02-networkConstruction-auto.RData")
4关联共识模块与雌性小鼠特定模块
4.1 加载雌性小鼠特定网络及模块
不知道大家还记不记得单纯在雌性小鼠中构建的网络和模块,这里我们需要再次load进来。😘
load("./FemaleLiver-02-networkConstruction-auto.RData")
4.2 整理一下
为了避免冲突,我们修改一下变量名。🐵
femaleLabels <- moduleLabels
femaleColors <- moduleColors
femaleTree <- geneTree
femaleMEs <- orderMEs(MEs, greyName = "ME0")
4.3 加载共识网络和模块
然后我们再把之前一步法建立的网络和模块加载进来。🥳
load("Consensus-NetworkConstruction-auto.RData")
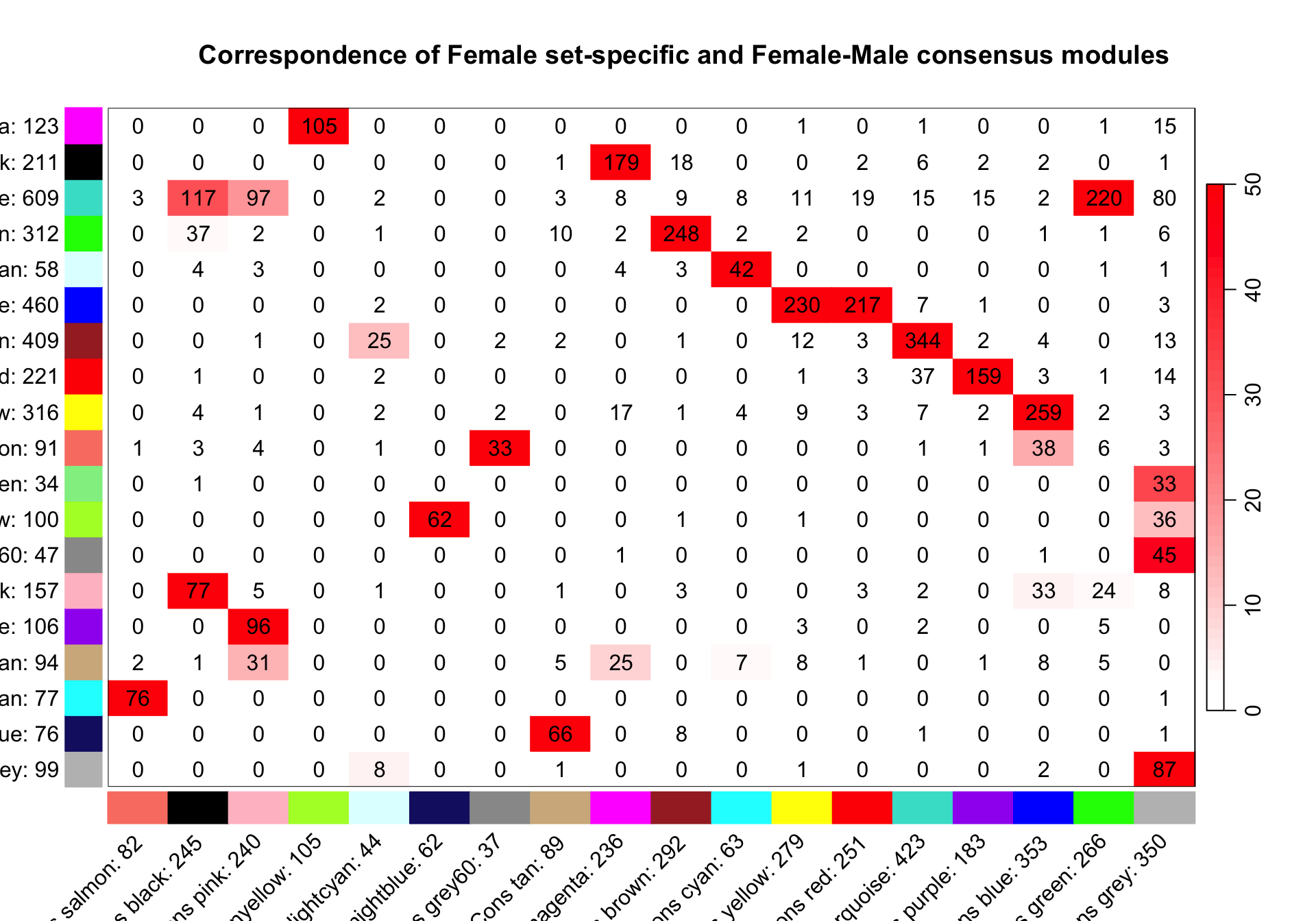
5计算Overlap部分
共识网络分析结果包括consMEs、moduleLabels、moduleColors 和consTree等。😉
我们现在准备好将雌性小鼠模块与共识模块相关联,然后计算一下overlap的部分,并使用超几何检验为每个overlap分配一个p值。😜
5.1 按模块eigengenes出现的顺序对模块进行label
femModuleLabels <- substring(names(femaleMEs), 3)
consModuleLabels <- substring(names(consMEs[[1]]$data), 3)
5.2 将数字标签转换为颜色标签
femModules <- labels2colors(as.numeric(femModuleLabels))
consModules <- labels2colors(as.numeric(consModuleLabels))
nFemMods <- length(femModules)
nConsMods <- length(consModules)
5.3 计算p值和相应计数的表
pTable <- matrix(0, nrow = nFemMods, ncol = nConsMods)
CountTbl <- matrix(0, nrow = nFemMods, ncol = nConsMods)
5.4 进行配对比较
for (fmod in 1:nFemMods)
for (cmod in 1:nConsMods)
{
femMembers = (femaleColors == femModules[fmod])
consMembers = (moduleColors == consModules[cmod])
pTable[fmod, cmod] = -log10(fisher.test(femMembers, consMembers, alternative = "greater")$p.value)
CountTbl[fmod, cmod] = sum(femaleColors == femModules[fmod] & moduleColors ==
consModules[cmod])
}
5.5 p值及可视化参数设置
pTable[is.infinite(pTable)] = 1.3*max(pTable[is.finite(pTable)])
pTable[pTable>50 ] = 50
femModTotals <- apply(CountTbl, 1, sum)
sizeGrWindow(10,7 )
par(mfrow=c(1,1))
par(cex = 1.0)
par(mar=c(8, 10.4, 2.7, 1)+0.3)
5.6 可视化
这里颜色代表的是p值哦。😜
labeledHeatmap(Matrix = pTable,
xLabels = paste(" ", consModules),
yLabels = paste(" ", femModules),
colorLabels = T,
xSymbols = paste("Cons ", consModules, ": ", consModTotals, sep=""),
ySymbols = paste("Fem ", femModules, ": ", femModTotals, sep=""),
textMatrix = CountTbl,
colors = greenWhiteRed(100)[50:100],
main = "Correspondence of Female set-specific and Female-Male consensus modules",
cex.text = 1.0, cex.lab = 1.0, setStdMargins = F)

点个在看吧各位~ ✐.ɴɪᴄᴇ ᴅᴀʏ 〰
📍 🤣 chatPDF | 别再自己读文献了!让chatGPT来帮你读吧!~
📍 🤩 WGCNA | 值得你深入学习的生信分析方法!~
📍 🤩 ComplexHeatmap | 颜狗写的高颜值热图代码!
📍 🤥 ComplexHeatmap | 你的热图注释还挤在一起看不清吗!?
📍 🤨 Google | 谷歌翻译崩了我们怎么办!?(附完美解决方案)
📍 🤩 scRNA-seq | 吐血整理的单细胞入门教程
📍 🤣 NetworkD3 | 让我们一起画个动态的桑基图吧~
📍 🤩 RColorBrewer | 再多的配色也能轻松搞定!~
📍 🧐 rms | 批量完成你的线性回归
📍 🤩 CMplot | 完美复刻Nature上的曼哈顿图
📍 🤠 Network | 高颜值动态网络可视化工具
📍 🤗 boxjitter | 完美复刻Nature上的高颜值统计图
📍 🤫 linkET | 完美解决ggcor安装失败方案(附教程)
📍 ......
本文由 mdnice 多平台发布