期刊:Cell Host Microbe
影响因子:31.316
发表时间:2022.7
- 一、摘要 -
人类肠道病毒组通常被称为肠道微生物组的“暗物质”,仍未得到充分研究。了解不同人群肠道病毒组的组成和变化对于探索其对人类健康的影响至关重要。人类肠道病毒组研究揭示了肠道病毒的高度遗传多样性和各种功能潜力。本综述总结了最近可用的四个人类肠道病毒组数据库,并讨论了它们的特征、构建过程和挑战,旨在为研究人员在选择数据库时提供参考。本综述还提出了对病毒种群进行分类的“最佳实践”。
- 二、背景介绍 -
粪便中人类肠道病毒浓度为109-10个病毒颗粒(VLPs)/g,是肠道微生物群中不可忽视的组成部分,具有高丰度、时间持久性和显著的多样性。虽然存在真核病毒,但人类肠道病毒组主要由噬菌体构成。最近的研究表明,病毒组可能在人类肠道菌群的生态过程中发挥重要作用。在炎症性肠病、糖尿病、高血压、艾滋病、结直肠癌和急性营养不良中,已报道肠道病毒组的疾病特异性改变。
肠道病毒组研究一般有两种测序策略:VLP宏基因组学(也称为病毒宏基因组学)和整体宏基因组学。
基于病毒RefSeq数据库的分析是直接将宏基因组测序reads与包含已知肠道相关病毒的公共数据库对比,导致对肠道病毒群组成的描述较差和不完整。因此,宏基因组测序和重新组装相结合被认为是揭示肠道噬菌体种群多样性和促进新病毒基因组发现的关键方案。过去几年里,一些人类肠道病毒库相继发表,这些数据库极大地扩展了人们对人类肠道病毒基因组的认识,并提供了丰富的注释信息。
然而,这些数据库之间可区分的特征和构建方法有差异,这导致了两个关键问题:①如何选择合适的数据库;②如何选择适当的参考程序。本综述讨论了肠道病毒数据库构建的差异和影响,并预测了未来研究中仍需解决的挑战。
- 三、人肠道病毒体的特征数据库 -
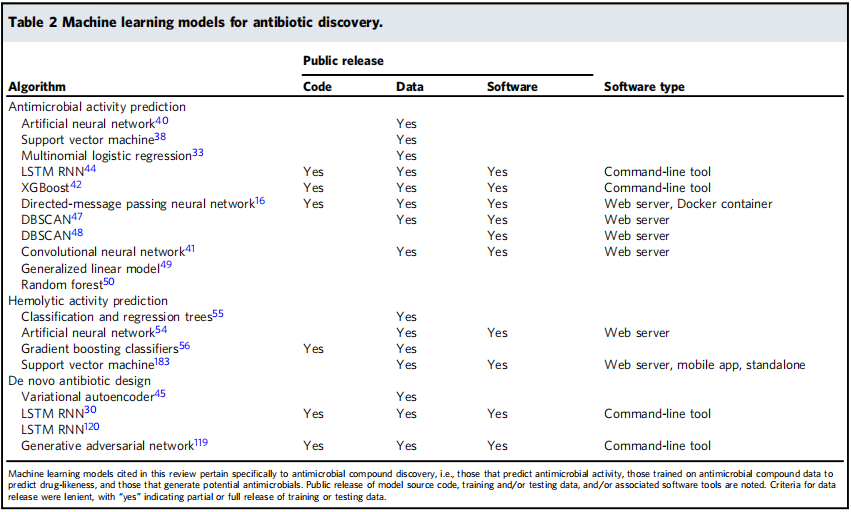
对过去2年发表的大规模宏基因组数据集建立的4个肠道病毒组数据库进行描述。
1)肠道病毒数据库(GVD)是基于2,697个人类肠道宏基因组数据,获得33,242个病毒种群(vOTUs),并记录了病毒多样性在健康个体的整个生命周期中的变化。
2)Cenote人病毒组数据库(CHVD)分析人不同部位(肠道、口腔、鼻子、皮肤和阴道)的5996个宏基因组样本测序数据,获得45033个非冗余的vOTUs,并发现了2200多个病毒类群与帕金森病和肥胖等几种慢性疾病的关联。
3)宏基因组肠道病毒数据库 (MGV) 是基于已发表的11,810 个人类粪便宏基因组测序数据,对病毒基因组进行大规模鉴定, 获得54,118 个vOTUs,这些基因组极大地扩展了肠道微生物组中 DNA 病毒的已知多样性,并提高了对宿主-病毒关联的了解。
4)肠道噬菌体数据库(GPD)是通过调查28,060个人类肠道宏基因组形成的一个更广泛的人类病毒库,其中包含142,809个非冗余肠道噬菌体基因组。GPD将噬菌体与特定的细菌宿主联系起来,并披露了人类肠道病毒体的全球分布特征。
这四项研究共同证明了人类肠道病毒体的高度多样性。随着样本量的增加,识别到的vOTU数量也在增加(图1A),这意味着目前发布的肠道病毒库并未达到饱和。
表1 4个人类肠道病毒库的情况

维恩图(图1B)显示了用于构建这四个肠道病毒库研究的重叠情况。78.8%的GVD研究和24.0%的CHVD研究采用VLP宏基因组学策略(图1C)。
据报道,地理、饮食、遗传和药物等宿主因素会塑造人类肠道病毒体。在一项针对中国人群的肠道病毒体研究中,发现地理对人类肠道病毒体变异的影响最为显著。本综述分析了4个数据库样本的地理分布(图1D),GPD从大洋洲收集的样本为3432个,远远超过了MGV、CHVD和GVD,这意味着GPD更适合大洋洲肠道病毒群的研究。

图1 四个肠道病毒库样本数量、已发表的研究以及样本的地理组成
四、病毒数据库的构建流程
通过从宏基因组数据集中组装病毒基因组,可以建立一个更全面的病毒数据库。上述这四个肠道病毒数据库从28,000个样本中收集到肠道病毒组,为肠道病毒的研究奠定良好基础。然而,四个数据库在执行类似程序的方式上有差异,因此,需要一种对人类肠道病毒体进行编目的标准化过程。
本综述将这些过程分为五个部分:reads组装、病毒contig识别、基因组质量评估、分类学注释和细菌宿主分配。结合每项研究的优点,提出了一个肠道病毒体编目的“最佳实践”实现。
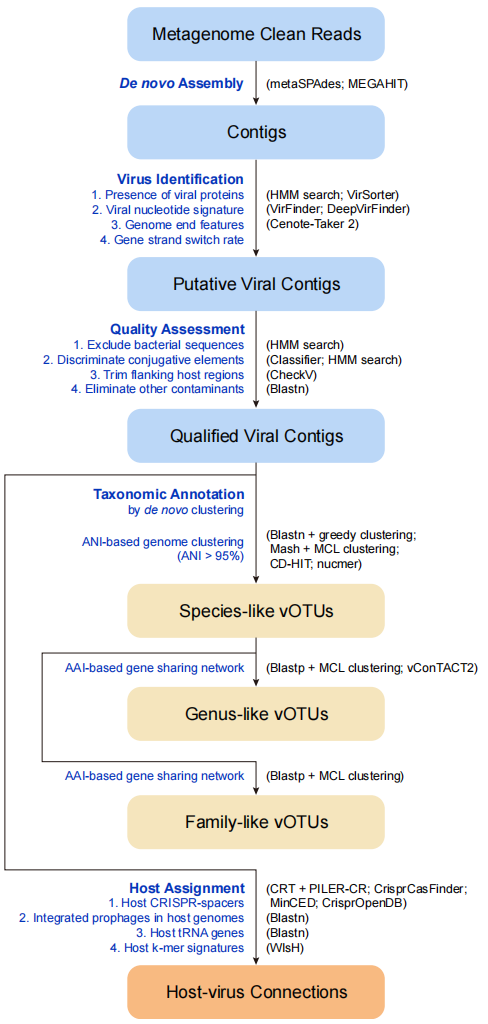
图2 肠道病毒组编目的工作流程
五、宏基因组组装、病毒识别和质量评估
人类肠道菌群的大部分宏基因组测序数据来自细胞生物,为了解决这一问题,肠道病毒数据库的研究包括三个过程:宏基因组测序数据的组装、推定病毒序列的识别、病毒基因组的质量控制和评估。
组装:肠道病毒的高度多样性、个体特异性、可变丰度以及整合前噬菌体和宿主基因组序列之间的模糊边界等特征阻碍了病毒基因组的精确组装:MEGAHIT和metaSPAdes是目前病毒库研究中常用的两种方法。MEGAHIT在组装大型复杂宏基因组数据时具有更快的速度和更少的计算机内存消耗,而metaSPAdes的特点是对计算资源的需求更高,但在获得较长的contigs时具有更好的性能。
病毒序列的识别:肠道病毒缺乏一种通用的标志基因,这里讨论的研究应用了各种复杂的方法来识别病毒,包括:(1)VirSorter软件是使用隐马尔可夫模型(HMM)搜索病毒蛋白家族的存在;(2)VirFinder或DeepVirFinder是利用病毒核苷酸特征,如k-mer频率和GC skew值;(3)基因组末端特征,如直接末端重复序列和反向末端重复序列;(4)同一链上的多个相邻基因。多种方法联合使用。
对假定病毒序列进行质控去除假阳性序列,通常是基于以下几种方法进行的:
(1)通过HMM搜索细菌蛋白家族排除细菌序列;
(2)使用机器学习分类器或HMM搜索将噬菌体与结合移动元件区分;
(3)使用CheckV或Cenote-Taker 2修剪前噬菌体的宿主侧翼序列;
(4)去除人类、动物序列;
(5)消除已知的污染物。
去除假阳性序列后,通过CheckV自动化流程估计基因组完整性来评估剩余病毒contigs的质量。如表1所示,三个数据库中病毒的完整度都不高,这表明目前病毒基因组数据库中的基因组很大程度上是碎片化的。
总之,以上三个步骤在GPD、MGV、CHVD和GVD研究中是相似的,但每一项研究对病毒鉴定和质量控制都不尽相同,每个研究使用的不同标准是互补的,未来的研究可能通过综合考虑所有这些标准来获得更高的特异性和更有利的表现。
- 六、细菌寄主分配 -
鉴定噬菌体的宿主范围对于理解肠道微生态至关重要,包括以下内容:
(1)噬菌体动力学和进化是如何被重塑的;
(2)细菌生长和代谢是如何重新连接的;
(3)如何促进水平基因转移(HGT);
(4)噬菌体相互作用如何影响人类健康。
计算方法可分为依赖序列对比和不依赖序列比对。依赖对比方法通过筛选与噬菌体匹配的CRISPR间隔区来进行。以此为原理的工具包括CRISPRDetect和CRISPRCasFinder。然而,大多数新组装的噬菌体序列与任何参考数据库都不匹配,并且只有大约40%的细菌携带CRISPR-Cas系统。
不依赖于序列比对的工具可以增加宿主预测百分比。基于基因组寡核苷酸k-mer频率的机器学习的工具,其预测准确率在28%~81%之间(属水平)。不依赖于序列对比的工具显示出相对较低的准确性,并且预测通常限于属水平及以上。
GPD、MGV、CHVD和GVD研究均采用了依赖序列比对方法以及CRISPR序列分配宿主(图2)。MGV内81%的噬菌体注释到宿主,其次是CHVD内69%、GVD内42%和GPD内29%(表1)。通过检查数据库构建的程序,研究者总结出两个可能对宿主预测性能至关重要的因素。首先,宿主分配很大程度上取决于CRISPR序列数据库,特别是数据库的大小和来源。
总之,依赖对比方法的宿主预测的性能主要受间隔数据库的大小和来源以及对齐参数设置的影响。研究人员可以结合结合多个CRISPR间隔区数据库,以提高宿主预测的灵敏度,根据研究的目的选择适当的对齐参数。还可以改进机器学习模型和输入特征,如phisdetector和VirHostMatcher-Net使用集成特征来提高预测精度。总之,必须整合不依赖比对和依赖比对的方法,在提高宿主预测的灵敏度的同时保证准确性。
- 七、总结与展望 -
本综述总结了四个肠道病毒数据库的特点,有助于后续研究选择更合适的数据库。此外,研究者发现涉及的宏基因组数据集越多,识别出的物种样vOTU数量越多,表明目前的数据库仍然没有达到肠道病毒多样性的饱和。通过普通病毒宏基因组测序鉴定ssDNA、ssRNA和dsRNA噬菌体存在局限性。综述总结了肠道病毒组分类时涉及的典型程序,提出了一种考虑到每项研究优点的“最佳实践”方法。希望这一最佳实践方法将对计划建立或更新病毒数据库的研究人员具有指导意义和信息价值。
新病毒的注释似乎更具挑战性,在属和科的水平上,关于阈值和聚类算法还没有达成共识。仍需要开发一种可行和稳健的噬菌体系统发育方法。最后,研究者注意到在四个病毒组数据库中细菌宿主指定的噬菌体的比例变化很大,受所使用的CRISPR间隔区数据库大小和错配设置的影响,增加间隔区序列比对中的错配数目会显著降低细菌宿主预测的准确性。
除了上述的进展和挑战,目前的研究对病毒进行分类仅仅是揭开肠道病毒奥秘的开始。类似于研究肠道细菌组与人类健康之间因果关系的策略,未来应开展一系列全病毒关联研究。鉴于病毒基因组表现出独特的特征,并且噬菌体系统发育的局限性,现有的微生物组分析工具可能不适用于病毒范围内的关联分析。因此,应广泛努力开发专门用于病毒组研究的分析工具包,如高效的病毒组图谱分析和噬菌体-宿主动态相互作用分析。
参考文献
Advances and challenges in cataloging the human gut virome