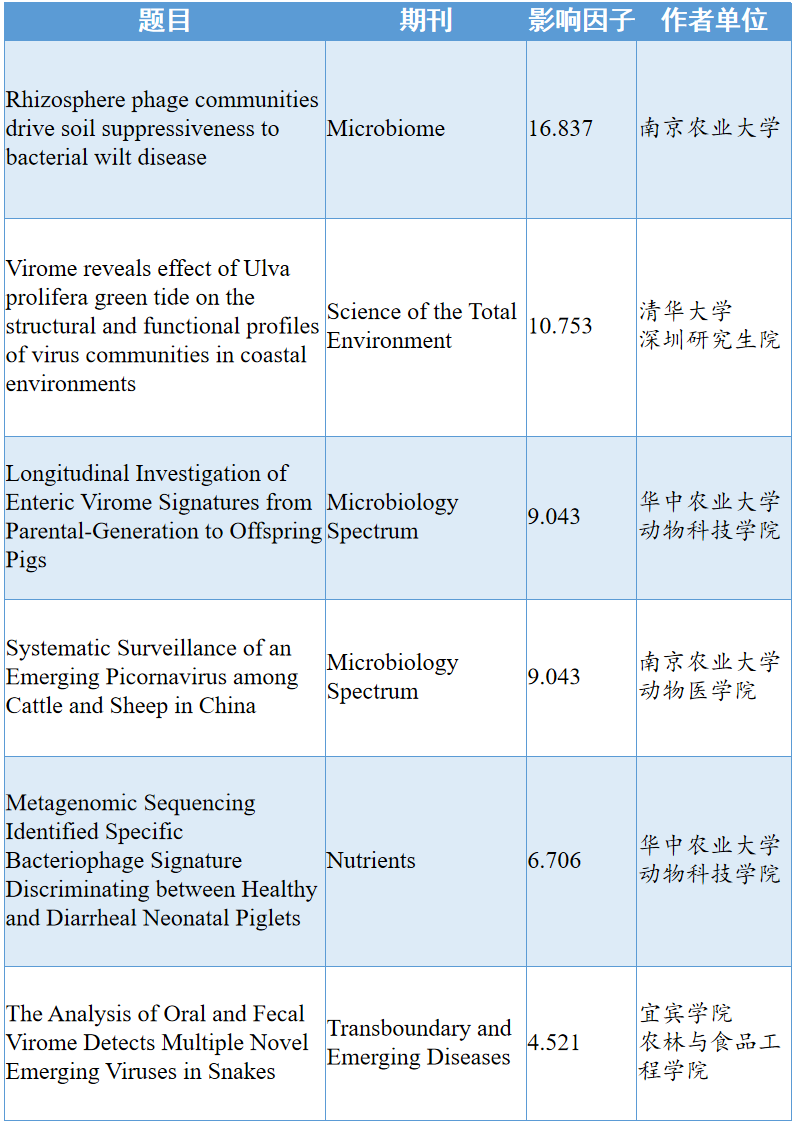
文章目录
- 建筑环境中的人为微生物耐药性
- 热心肠日报
- 摘要
- 背景
- 结果
- 限制与微生物多样性减少相关
- 附表1. 基于单个宏基因组样品blastx比对NCBI NR估计Alpha多样性
- 图1. 宏基因组数据物种和功能香农多样性指数组间比较
- 附图1. 16S物种香农多样性指数组间比较
- 环境差异与微生物组相关
- 图2. 不同环境的聚类
- 附图2. 组内距离分布
- 附图3. 域组成
- 附图4. 门组成
- 附图5. 物种组成
- 附图6. 限制环境中独特的物种分类
- 附图7. 核心微生物组
- 图3. NMDS分析样本组和环境因子
- 基因组水平上功能明显改变
- 附表2. 每类样品中分箱(Bin)的结果统计
- 图4. 基因组概述
- 附图8. 革兰氏阳性细菌
- 附图9. 革兰氏阴性细菌
- 附图10. 致病潜力
- 附图11. 潜在胁迫耐受
- 附图12. 受限不同组特异的16S物种类别
- 附表3. 不同环境和物种的泛和核心基因组
- 附图13. 功能组成柱状图
- 附图14. 功能组成放射图
- 附图15. 独特的功能
- 附图16. 物种和功能分别主坐标轴分析
- 附图17. 主要的基因组功能
- 抗生素抗性组存在差异
- 图5. 抗性特征的多样性分析
- 附图18. 基因组和质粒的抗性组进行PCoA
- 图6. 基因组和质粒的抗性网络
- 图7. CARD高级分类和药物分类
- 讨论
- 方法
- 环境参数和研究设计
- 附图19. 非限制的建筑环境(UB)
- 附图20. 限制的建筑环境-ICU
- 附图21. 限制的建筑环境-洁净室
- 附表4. 用于采样建筑环境中各种表面和使用的清洁和消毒试剂
- 附表5. 包括奥地利格拉茨国立医院ICU中消毒剂种类和使用时间
- 表1. 环境参数
- 取样过程
- 取样处理和DNA提取
- 定量测量
- 宏基因组测序
- 16S rRNA基因扩增与测序
- 附表6. 本研究中使用的引物
- 实验对照
- 生信分析
- 附表7. 宏基因组数据集统计
- 附表8. 宏基因组组装的重叠群(contig)和骨架(scaffolds)统计
- 附表9. 生信分析软件参数
- 统计信息
- 附表10. 16S数据上应用的统计方法
- 附表11. 对PICRUSt结果统计的方法和显著性
- 附表12. 16S数据的样本统计信息
- 猜你喜欢
- 写在后面

主编按:第一次看到此文的推送是因为它引用了我参与的QIIME2,当时并没有引起我多大注意,过几天一位同行发给我此文请教其中的分析思路。仔细一读,发现它的实验设计、分析非常全面。极其适合作为纯分析文章的图表整理学习模板,故精读此文,供同行学习与讨论。以下全文共有2.3万字,7个主图,1个表,21个附图,12个附表,掌握以下内容,应对一般的扩增子+宏基因组分析项目无忧,更有机会冲击高分文章。一起学习吧!
建筑环境中的人为微生物耐药性
Man-made microbial resistances in built environments
Nature Communication, [12.353]
https://doi.org/10.1038/s41467-019-08864-0
Published: 27 February 2019
第一作者:Alexander Mahnert1
通讯作者:Alexander Mahnert1 alexander.mahnert@gmail.com
合作作者: Christine Moissl-Eichinger2,3, Markus Zojer4, David Bogumil5, Itzhak Mizrahi5,
Thomas Rattei 4, José Luis Martinez6 & Gabriele Berg1,3
1 奥地利格拉茨科技大学内科医学系 (Institute of Environmental Biotechnology, Graz University of Technology, Petersgasse 12/I, Graz 8010, Austria)
2 奥地利格拉茨科技大学环境生物技术研究所 (Department of Internal Medicine, Medical
University Graz, Auenbruggerplatz 2, Graz 8036, Austria.)
3 奥地利格拉茨科技生物技术医学中心 (BioTechMed Graz, Mozartgasse 12/II, Graz 8010, Austria)
4 维也纳大学,微生物学和生态系统科学系,计算系统生物学, (Division of Computational Systems
Biology, Department of Microbiology and Ecosystem Science, University of Vienna, Althanstrasse 14, Vienna 1090, Austria)
5 以色列内盖夫本古里安大学自然科学学院命科学系 (Department of Life Sciences,
Faculty of Natural Sciences, Ben-Gurion University of the Negev, Box 653, Beer-Sheva 84105, Israel)
6 马德里,卡莱达尔文,CSIC,生物技术中心
(Centro Nacional de Biotecnologia, CSIC, Calle Darwin 3, Madrid 28049, Spain.)
热心肠日报
https://www.mr-gut.cn/papers/read/1076375825
Nature子刊:恢复菌群多样性或能降低耐药性
创作:方芳 审核:小肠君 03月11日
原标题:建筑环境中的人为微生物耐药性
- 医院的ICU病房、清洁室、更衣室更封闭,消毒措施更严格,这种人为的清洁手段可导致环境菌群多样性下降,主导细菌由革兰氏阳性菌转变为革兰氏阴性菌;
- 相比其他建筑环境,相对封闭、消毒措施严格的临床环境中,环境菌群具有不同的功能基因和耐药基因组,细菌对极端环境的耐受性更强、致病性更强,耐药基因的多样性更高;
- 菌群多样性的丧失、菌群功能的变化与耐药性增加有关;
- 为减轻抗生素耐药性,可能需要恢复环境中的菌群多样性。
主编评语:人为因素对环境菌群的耐药性具有重要影响。《Nature Communications》近期发表研究,比较临床环境表面和其他建筑环境的菌群,既耐药基因组,更封闭,消毒措施更严格的临床环境中菌群多样性下降、耐药基因的多样性增加,指示菌群的高多样性可能有助于维护有益菌群、降低耐药基因丰度。该结果对于临床管控抗生素耐药性具有重要参考价值。
摘要
抗菌素耐药性是对全球公共卫生的严重威胁,但微生物控制(microbial control)对微生物组及其相关耐药性的影响知之甚少。我们将临床环境表面的微生物组与其他建筑环境进行比较。利用最先进的宏基因组学、基因组和质粒重建方法,发现增加限制和清洁与微生物多样性的丧失以及从革兰氏阳性菌(如放线菌门和厚壁菌门)向革兰氏阴性菌(如变形菌门)的转变有关。此外,与其他建筑环境相比,高度维护建筑环境的微生物组具有不同的抗性,并且抗性基因的多样性更高。我们的研究结果强调,微生物多样性的丧失与耐药性的增加以及在某些建筑环境中实施恢复细菌多样性的必要性。
Antimicrobial resistance is a serious threat to global public health, but little is known about the effects of microbial control on the microbiota and its associated resistome. Here we compare the microbiota present on surfaces of clinical settings with other built environments. Using state-of-the-art metagenomics approaches and genome and plasmid reconstruction, we show that increased confinement and cleaning is associated with a loss of microbial diversity and a shift from Gram-positive bacteria, such as Actinobacteria and Firmicutes, to Gram-negative such as Proteobacteria. Moreover, the microbiome of highly maintained built environments has a different resistome when compared to other built environments, as well as a higher diversity in resistance genes. Our results highlight that the loss of microbial diversity correlates with an increase in resistance, and the need for implementing strategies to restore bacterial diversity in certain built environments.
背景
Introduction
与抗生素抗性细菌感染相关的发病率和死亡率的增加是当今人类面临的主要全球性威胁之一。抗菌素耐药性(Antimicrobial resistance,AMR)被认为是一个真正的健康危机,必须在几个方面得到有力解决。其中大部分与环境中的人类行为有直接相关。近年来,AMR在环境方面的研究集中在畜牧业、废水处理和医院环境等方面。然而在这些研究中,通常人类在其中度过大部分生活时间的其他建筑环境(如私人住宅和工作场所)往往被忽视,尽管它们与AMR的出现和传播有潜在的相关性。一个例外是由LAX和同事进行的研究,他们不仅调查了AMR在医院的设施,也调查了私人住宅。
The increased morbidity and mortality rate associated with infections by antibiotic-resistant bacteria is one of the main global threats human kind has to face nowadays. Antimicrobial resistance (AMR) is recognized as a real health crisis that has to be forcefully tackled on several fronts1. Most of these fronts are directly linked to human behavior in the environment2. In recent years, research has been focused among others on the environmental dimension of AMR especially in livestock farming, waste water treatment, and in hospital settings. However, other built environments in which people commonly spend most of their lives (e.g., private homes and workplaces) have been often neglected in these studies, despite their potential relevance for the emergence and spread of AMR. An exception is the study by Lax and coworkers, who investigated AMR not only in a hospital setting3, but also in private homes4.
根据科学界的共识标准原则,我们比较了临床环境和其他建筑环境中表面AMR的相关性。我们重点研究了人为因素对建筑物影响的三个主要方面:
- 占用率和通道类型;
- 房间使用情况;
- 可能改变微生物组的人类活动,如一般的微生物控制和清洁。
由于先前的研究已经表明,微生物群落和抗性结构与人类在其环境中的活动有关,因此我们有兴趣通过对抗性的深入分析,了解微生物控制和建筑限制如何影响居住微生物群落的组成和功能,及可移动的遗传元件(mobile genetic elements)。
In line with such demands by the scientific community2,5, we compared associations of AMR between surfaces in clinical settings and other built environments. We focused on three main aspects of anthropogenic influences on buildings: (1) occupancy and type of access, (2) room’s usage, and (3) human activities that may alter the microbiota like microbial control and cleaning, in general. Since prior studies have already indicated that microbial community and resistome structures correlate with human actions in their environment3,4,6,7, we were interested in learning how microbial control and building confinement affect the composition and functional capabilities of the residing microbiome with an in-depth analysis of the resistome and its mobile genetic elements.
为此,我们定义了一组模型量化差异的建筑环境,这些环境在人为影响的等级上有所不同,包括微生物控制、清洁和与外界交换程度。一方面,我们调查了不同的自然无限制建筑(unrestricted buildings, UBs)和受周围户外环境(包括植物)影响程度较高的房屋,这些建筑位于农村地区。另一方面,我们对控制性建筑环境(controlled built environments, CBs)进行了抽样,随着微生物限制和清洁操作水平的不断提高,从重症监护室(intensive care units, ICU)到郊区的航天器装配洁净室设施(spacecraft assembly cleanroom facilities)。所有样本都得到了丰富的环境元数据集合的支持,以将微生物组分和功能与环境参数相关联。这种独特的研究设计得到了一种新的取样方法的进一步支持,即使是在低生物量的环境中,也能获得深度测序的宏基因组文库。此外,我们还进行了以基因组为中心的最新生物信息学分析(Binning和抗性基因注释),以阐明其基因组背景下的抗性特征。
For this purpose, we defined a set of model built environments, which differ in their grade of anthropogenic influences, including microbial control, cleaning, and access. On one hand, we investigated different naturally unrestricted buildings (UBs) and houses with a high level of influence from the surrounding outdoor environment, including plants, in a rural setting. On the other hand, we sampled controlled built environments (CBs) with an increasing level of microbial confinement and cleaning operations from intensive care units (ICUs) to spacecraft assembly cleanroom facilities in urban areas. All samples were supported by a rich collection of environmental metadata to correlate compositions and functions of the microbiome with environmental parameters. This unique study design was further supported by a new sampling methodology to acquire deeply sequenced shotgun libraries even from low-biomass environments. In addition, a state-of-the-art genome centric bioinformatics analysis8 was conducted to elucidate resistance features in their genome context.
以上这些新见解有助于模拟人类驱动影响建筑内部环境微生物组及其相关抗性的过程,并改善我们对在建筑环境中保存或最终设计微生物组可能性的评估。
These new insights are useful to model human-driven processes affecting in-house microbiota and its associated resistome and to improve our assessments on the possibilities of preserving or, eventually, designing microbiomes in built environments.
结果
Results
限制与微生物多样性减少相关
Confinement correlates with reduced microbial diversity
UB和CB的对立结构伴随着显著的分类多样性损失(斯皮尔曼秩相关系数rho = -0.8783,P = 0.02131;单侧t检验:n = 9,t = -3.2,df = 2.6,P = 0.03)以及附表1)。相比之下,UB和CB之间的功能多样性保持平衡(根据SEED注释,10.8–11.1 H’;图1b)。由于ICU样本的多样性估计值较低(3.8 H’)和私人住宅的多样性较高(6.4 H’),16S rRNA序列分析显示CB(5.6 H’)和UB(7.2 H’)之间的差异更为明显(附图1)。在细菌丰度恒定(每平方米约106-107个16S rRNA基因拷贝数)的情况下观察到多样性估计的这些差异,完整细胞的部分变异性较高(每平方米约103-107个16S rRNA基因拷贝数)。然而,多样性估计与完整细胞的比例不相关(斯皮尔曼秩相关系数rho = 0.2,P = 0.4)。
Opposing structures of UB and CB were accompanied by a significant loss (Spearman’s rank correlation rho, correlation coefficient: -0.8783, P = 0.02131; one-sided t test: n = 9, t = -3.2, df = 2.6, P = 0.03) of taxonomic diversity (Shannon–Weaver indices: CB 7.2 H’, UB 8.8 H’) (Fig. 1a and Supplementary Table 1). In contrast, the functional diversity between UB and CB remained balanced (10.8–11.1 H’ according to SEED annotations; Fig. 1b). The analysis of 16S rRNA sequences showed even clearer differences between CB (5.6 H’) and UB (7.2 H’) due to lower diversity estimates for ICU samples (3.8 H’) and a higher diversity for private houses (6.4 H’) (Supplementary Fig. 1). These differences in diversity estimates were observed in the presence of constant bacterial abundances (~ 106–107 16S rRNA gene copies per m2), with a higher variability for the fraction of intact cells (~103–107 16S rRNA gene copies per m2). However, diversity estimates did not correlate with the proportion of intact cells (Spearman’s rank correlation rho, correlation coefficient: 0.2, P = 0.4).
附表1. 基于单个宏基因组样品blastx比对NCBI NR估计Alpha多样性
Supplementary Table 1: Alpha diversity estimates from single shotgun reads of the metagenomics dataset against the NCBI nr database using the blastX algorithm.

分为四列,为物种,物种(去除人源相关注释),KEGG和SEED数据库的结果,计算Shannon和Simpson指数。
图1. 宏基因组数据物种和功能香农多样性指数组间比较
采用小提琴图+箱线图展示 Shannon 多样性指数,建议方案可改为theme_bw更好看,更合理的方法数据不多可jitter添加散点,和统计(统计应该不显著),图注中要标明样本数量。

微生物多样性估计。根据对NCBInr的blastx搜索结果,用MEGAN进行计算。对只出现一次的reads进行过滤(去除未分配的reads),并进行归一化(随机和重复地将样本大小减至最小样本量)。在R中创建显示数据的核概率密度的小提琴图,包括一个带有中位数和四分位间距的方框(小提琴图+箱线图)。
a. 在CB(受限)和UB(无限制建造环境)中,香农微生物群落物种多样性估计的显著差异。(説显著,但看关不显著 。significant出现时,必须写P值和统计方法,否则图注不完整,但在正文中只提了明显不一致;不显著可有明显obvious、distinct clear,substantial等词形容不需统计)
b. 类似的香农多样性——在最高功能注释SEED数据库层级 (个体功能基因水平,5级)上对CB(受限)和UB(无限制建造环境)微生物功能的估计。(功能多样性无明显差异)
Microbial diversity estimates. Calculations were executed in MEGAN according to the results of the BLASTx searches against NCBInr. Data of single reads were filtered (unassigned reads were removed) and normalized (randomly and repeatedly subsampled to the smallest sample size). Violin plots showing the kernel probability density of the data, including a box with the median and the interquartile range, were created in R. a Significant differences of Shannon diversity estimates of microbial communities on species level in CB (confined) and UB (unrestricted built environments). b Similar Shannon diversity estimates of microbial functions on highest SEED levels (individual functional gene levels, level 5) in CB (confined) and UB (unrestricted built environments)
附图1. 16S物种香农多样性指数组间比较
Supplementary Figure 1: Diversity estimates

基于16S rRNA基因扩增子分析的受限CB和非受限UB建筑环境的多样性估计。扩增子的结果中,受限环境的多样性明显低。
Diversity estimates of confined and unrestricted built environments based on 16S rRNA gene amplicon analysis.
环境差异与微生物组相关
Environmental differences correlate with the microbiome
基于无权重的PCoA分析,公共建筑和公共房屋的宏基因组样本更相似。在UB和CB组间存在更大差异。此外UB内存在很小差异的群体结构(均值Bray–Curtis距离比0.71)比CB环境(bray–curtis均值距离0.82;图2和附图2)。注:是因为UB中有大量PMA处理的相近结果,导致组内差异小吧,相当于多了一批重复。
Shotgun metagenome samples from public buildings and public houses were more similar to each other than samples obtained from private houses according to Principal Coordinates Analysis (PCoA) ordinations and Unweighted Pair Group Method with Arithmetic Mean trees. Even greater dissimilarities were observed between samples from UB and CB. Moreover, 16S rRNA-based population structure indicated lower dissimilarities for UB (mean Bray–Curtis distance 0.71) than for CB environments (mean Bray–Curtis distance 0.82; Fig. 2 and Supplementary Fig. 2).
图2. 不同环境的聚类

不同构建环境类型之间的连接。基于不同微生物群落的UPGMA树(算术平均树的无权重对组法)被分析到物种水平。根据对NCBInr的blastx搜索结果,使用MEGAN进行计算。对单次读取的数据进行过滤(去除未分配的读取),并进行归一化(随机和重复地将样本大小减至最小)。柱环境色标:蓝色(洁净室设施);红色(重症监护室);深绿色(公共建筑);浅绿色(公共房屋);黄色(私人房屋)
Connection between different built environment types. UPGMA tree (Unweighted Pair Group Method with Arithmetic Mean tree) of sampled built environments based on different microbial communities resolved to species level. Calculations were executed with MEGAN according to the results of the BLASTx searches against NCBInr. Data of single reads were filtered (unassigned reads were removed) and normalized (randomly and repeatedly subsampled to the smallest sample size). Color code for column environment: blue (cleanroom facility); red (intensive care unit); dark green (public buildings); light green (public houses); yellow (private houses)
附图2. 组内距离分布
Supplementary Figure 2: Distance estimates

基于BC距离的16S扩增子数据估计CB和UB环境的距离
Bray-Curtis distance estimates of confined and unrestricted built environments based on 16S rRNA gene amplicon analysis
即使在超界层面上,不同种类的取样建筑环境也可以通过宏基因组的不同组成来描述(附图3,注:宏基因组跨多个生物界测序,包括细菌、真菌、原生动物、病毒等,可以从界层面比例讨论)。因此,从UB(99%细菌,~ 1%真核生物)到CB(细菌:洁净室~ 69%及其更衣区~ 85%;ICU ~ 55%)的细菌与真核生物(主要是分配给人类的序列)的比例显著下降(单侧t检验:n = 9, t = 3.4, df = 2.0, P = 0.04)。对于古生菌,也可以观察到类似的模式,但并不显著(单侧t检验: n = 9, t = 1.9, df = 2.0, P = 0.1)。CB和UB之间的病毒痕迹不太明显,但在ICU和公共场所环境中的相对丰度最高。明显的差异继续存在于更高的分类水平(附图4门水平和附图5物种水平):在门水平上,公共建筑和公共房屋主要由放线菌(高达50%)和变形杆菌(约21%)的序列构成。在私人住宅中,厚壁菌门的比例上升到55%。同样,用一叠氮丙啶(PMA,来自Nature Microbiology 中 可去除Relic DNA,即死亡或暴露的DNA)掩盖受损细胞的DNA后,厚壁菌门的比例也较高(厚壁菌门有大量芽孢菌类Bacilus,可能为休眠状态,不容易被PMA结合)。在CB中,细菌门的可分类率降低,多细胞生物的比例增加,不可分配的序列增加(在洁净室高达62%)。此外,假单胞菌、卟啉单胞菌、丙酸杆菌和嗜氯菌可通LEfSe(效应大小的线性鉴别分析)确定为CB中的显著鉴别特征(附图6)。除这些细菌类群外,病毒序列(如人疱疹和乳头瘤病毒)和节肢动物(如蜱虫Trombidiformes和前气门亚目螨 prostigmata等)和昆虫(如虱子,如Liposcelis bostrychophila和 Blattella germanica蟑螂)也被定义为CB的鉴别特征。
Different categories of sampled built environments could be characterized by distinct compositions of the metagenomic reads even on the superkingdom level (Supplementary Fig. 3). Hence, proportions of bacteria vs eukaryota (mainly sequences assigned to humans) decreased significantly (one-sided t test: n = 9, t = 3.4, df = 2.0, P = 0.04) from UB (~ 99% bacteria, ~ 1% eukaryota) towards CB (for bacteria: cleanroom ~ 69% and its gowning area ~ 85%; ICU ~ 55%). A similar pattern could be observed for archaea, although not significant (one-sided t test: n = 9, t = 1.9, df = 2.0, P = 0.1), with higher counts (~ fourfold) in CB. Traces of viruses were less apparent between CB and UB, but showed highest relative abundances in the ICU and in the environment of public houses. Clear differences continued into higher taxonomic levels (Supplementary Fig. 4 and Supplementary Fig. 5): on the phylum level, public buildings and public houses were dominated by sequences of Actinobacteria (up to 50%) and Proteobacteria (~ 21%). In private houses, the proportion of Firmicutes raised up to 55%. Likewise, the proportion of Firmicutes was also higher after masking the DNA of compromised cells with propidium monoazide (PMA). In CB, the prevalence of bacterial phyla was reduced and proportions of multicellular organisms and not assignable sequences increased (up to 62% in the cleanroom). Furthermore, Pseudomonas, Porphyromonas, Propionibacterium, and Prochlorococcus could be identified as significant discriminative features (Supplementary Fig. 6) in CB by LEfSe (linear discriminant analysis of the effect size) analysis. Besides these bacterial taxa, also viral sequences (e.g., human herpes and papillomavirus) and assignments to arthropods (e.g., mites like Trombidiformes and Prostigmata) and insects (e.g., lices such as Liposcelis bostrychophila and cockroaches like Blattella germanica) were defined as discriminative features for CB.
附图3. 域组成
Supplementary Figure 3: Domain profile

序列采用BLASTx对NCBI NR的域分类,来自MEGAN排除末分类并进行数据标准化。
Single reads BLASTx (rapsearch and diamond) vs. NCBI nr. superkingdom level (derived from MEGAN, excluding unassigned reads, normalized data set).
附图4. 门组成
Supplementary Figure 4: Phyla profile


序列采用BLASTx对NCBI NR的门水平分类,来自MEGAN排除末分类并进行数据标准化。
附图5. 物种组成
Supplementary Figure 5: Species profile


序列使用diamond采用BLASTx对NCBI NR的种水平分类,来自MEGAN排除末分类并进行数据标准化为百分比。
Space filling radial chart of taxa (species level, excluding unassigned reads, normalized, percentage) assigned (BLASTx NCBInr, diamond and rapsearch) to different built environments (MEGAN).
附图6. 限制环境中独特的物种分类
Supplementary Figure 6: Distinctive taxa of controlled built environments (CB)

用以下参数对CB(ICU、更衣区、洁净室)和UB(公共建筑、公共和私人住宅)构建环境的宏基因组单次测序的分类群(根据NCBI NR数据库)进行了LEfSe分析(LDA效应大小):按样本归一化到1百万(M),类间因素Kruskal-Wallis检验(alpha=0.01),成对亚类间Wilcoxon检验(alpha=0.01),LDA评分阈值(1.0),多类分析策略(all-against-all, more strict)。
LEfSe analysis (LDA effect size) on taxa (according to NCBInr database) of single reads from metagenomes of CB (ICU, gowning area, cleanroom) and UB (public buildings, public and private houses) built environments with the following parameters: per-sample normalization to 1M, factorial Kruskal-Wallis test among classes (alpha = 0.01), pairwise Wilcoxon test between subclasses (alpha = 0.01), threshold for the LDA score (1.0), strategy for multi-class analysis (all-against-all, more strict).
微生物组成的16S rRNA基因的核心OTU网络(附图7)。本分析展示了一个高比例的共有菌来自金黄色葡萄球菌、鲍曼不动杆菌,同时更大的重叠来自不受限制的样品从洁净室设施的建筑相比从ICU核心样品的环境。
注:此图不是共相关网络,而是不同菌与来源的对应关系,类似于维恩图,但是可用属大小代表菌的丰度,颜色代表共有、特有的类别。
The core 16S rRNA gene microbial profile was visualized in a core operational taxonomic unit (OTU) network (Supplementary Fig. 7). This analysis indicated a high proportion of shared OTUs assigned to Acinetobacter and Staphylococcus as well as a bigger overlap of samples from the cleanroom facility and unrestricted buildings compared to the core of samples from the ICU environment.
附图7. 核心微生物组
Supplementary Figure 7: Core microbiome

基于G检验的16S rRNA基因扩增子独立性核心OTU网络分解到属水平。边缘加权弹簧嵌入算法在Cytoscape软件中实现了可视化。节点大小反映了OTU的丰度。按线条宽度和透明度展示边权重。颜色指的是不同的采样建筑环境:洁净室设施(蓝色)、重症监护室(红色)、公共建筑、公共住宅、私人住宅(全部为绿色)。
Core OTU network based on G-tests for independence of 16S rRNA gene amplicons resolved to genus level. Edge-weighted spring embedded algorithms implemented in Cytoscape were used for visualizations. OTU abundance is reflected by node size. Edge weights by line widths and opacities. Colors refer to different sampled built environments: Cleanroom facility (blue), intensive care unit (red), public buildings, public houses, private houses (all in green).
为了将微生物群落组成与环境参数相关联,在16S rRNA基因组上进行了一项与欧氏距离相比的斯皮尔曼秩相关bioenv检验。这项bioenv分析显示,与温度、湿度和房间变量(如表面积、房间高度或房间体积)相比,样本与纬度、经度和海平面(最佳变量组合ρw = 0.9425)的相关性更高(最佳变量组合ρw = 0.7518)。这些相关性进一步被作为采样群落的非度量多维标度(NMDS)排序的向量(环境因子与PCoA主轴进行相关分析),以及每个采样类别的样品计算置信椭圆(图3)。这一排序显示,从私人住宅的瓷砖表面、公共住宅和公共建筑的卫生环境或ICU楼层和ICU工作场所与医疗器械的样本重叠处获取的样本具有明显的聚集。然而,由于混淆变量(见补充信息),微生物组与环境变量(如生物地理学或小气候)的关联不能得到进一步支持或区分。
To correlate microbial community composition with environmental parameters, a bioenv test with Spearman rank correlations compared to Euclidean distances was applied on the 16S rRNA gene profile. This bioenv analysis showed higher correlations of samples with latitude, longitude, and sea level (best variable combination ρw = 0.9425) than with temperature, humidity, and room variables, like the surface area, room height, or room volume (best variable combination ρw = 0.7518). These correlations were further visualized as vectors on an Non-metric multidimensional scaling (NMDS) ordination of the sampled communities together with calculated ellipses per sampling category (Fig. 3). This ordination showed distinct clusters for samples obtained from the surface of tiles in private houses, the sanitary environments in public houses and public buildings, or that ICU floors and ICU workplaces overlapped with samples from medical devices. However, associations of the microbiome with environmental variables like biogeography or microclimate could not be further supported or differentiated due to confounding variables (see Supplementary information)。
图3. NMDS分析样本组和环境因子

与采样建筑环境微生物组相关的环境变量。基于Bray–Curtis距离的16S rRNA基因扩增子的NMDS,叠加向量表示基于欧几里得距离的测量环境变量(bioenv)的Spearman相关性。柱环境色标:蓝色(洁净室设施);红色(重症监护室);深绿色(公共建筑);浅绿色(公共房屋);黄色(私人房屋)
variables associated with the microbiome of sampled built environments. NMDS of 16S rRNA gene amplicons based on Bray–Curtis distances with superimposed vectors representing Spearman correlations of measured environmental variables (bioenv) based on Eucledian distances. Color code for column environment: blue (cleanroom facility); red (intensive care unit); dark green (public buildings); light green (public houses); yellow (private houses)
总的来说,微生物群的组成非常明显,相关的元数据类别可以通过监督学习方法(随机森林分类和回归模型)进行预测。从CB或UB中提取的样本可预测,总准确度高达92%。同样,温度(R = 0.92, P = 4.8 × 10-5)、温度(R = 0.89, P = 3.3 × 10-4), 经度 (R = 0.95, P = 2.8 × 10-6), 和海平面 (R = 0.82, P = 3.3 × 10-3)可以很容易预测。微生物丰度(R = 0.63, P = 0.12)和各自的房间面积(R = 0.58, P = 0.24)不适合根据观察到的特征建立预测模型。
In general, the composition of the microbiome was so distinct that the associated metadata categories could be predicted by supervised learning methods (random forest classification and regression models). Samples from CB or UB could be predicted with a high overall accuracy of 92%. Likewise, numerical environmental parameters such as temperature (R = 0.92, P = 4.8 × 10-5), relative humidity (R = 0.89, P = 3.3 × 10-4), longitude (R = 0.95, P = 2.8 × 10-6), and sea level (R = 0.82, P = 3.3 × 10-3) could be easily predicted. Microbial abundances (R = 0.63, P = 0.12) and respective room areas (R = 0.58, P = 0.24) were not suitable to build predictive models from observed features.
基因组水平上功能明显改变
Changed functional capabilities were evident on genome levels
组装好的重叠群(contigs)和支架(scaffolds)可以组合成125个基因组草图(每个样本8-20个分箱)。大多数分箱基因组是从私人住宅样本中提取的,而只有少数基因组可以从ICU数据集中重建(附表2)。44个基因组草图的子集(占所有45%的重叠 群)的质量足以进行深入分析。这些分箱结合基因组的注释、复制活性和预测表型在CB或UB环境中具有显著的代表性(图4)。因此,根据IREP,CB(双侧双样本Kolmogorov-Smirnov检验:D = 0.68, P = 0.005)的复制率较低,10-75%的样本群体的复制事件范围为2-6次。根据表型分类算法(Phenotype Investigation with Classification Algorithms,PICA)的计算,在基因组和标记基因水平上可以预测出几个不同的表型(46个个体卡方检验,Bonferroni校正 P = 0.02)。因此,CB对烷烃降解、苯甲酸羟基化降解、胆碱生成三甲胺、T4和T6分泌系统以及基于thaxtomins的植物致病性具有显著的表型特征,而砷解毒和兼性厌氧菌对UB具有特异性。总的来说,革兰氏阳性细菌(P = 0.004)在UB中主要与碳水化合物和氨基酸代谢相关。相反,与毒力、疾病(P = 0.008)、防御(P = 5.2 × 10-5)和抵抗(P = 0.08)有关的革兰氏阴性细菌在CB中富集(P值由Kruskal–Wallis检验计算;附图8—11)。
Assembled contigs and scaffolds could be binned into 125 draft genomes (8–20 bins per sample). Most binned genomes were recovered from samples of private houses, while only a few genomes could be reconstructed from the ICU dataset (Supplementary Table 2). A subset of 44 draft genomes (representing 45% of all assembled contigs) were sufficient in quality for an in-depth analysis. Annotations, replication activity, and predicted phenotypes of these binned genomes were significantly representative for CB or UB environments (Fig. 4). Hence, according to iRep, replication rates were lower in CB (two-sided two-sample Kolmogorov–Smirnov test: D = 0.68, P = 0.005) and ranged from 2 to 6 replication events for 10–75% of the sampled population. According to Phenotype Investigation with Classification Algorithms (PICA), several distinct phenotypes could be predicted (46 individual chi square tests, Bonferroni correction P = 0.02) on genome and marker-gene levels. Therefore, significant phenotypic traits for CB covered alkane degradation, benzoate degradation by hydroxylation, trimethylamine production by choline, T4 and T6 secretion systems, and plant pathogenicity based on thaxtomins, while arsenic detoxification and facultative anaerobes were specific for UB. Overall, Gram-positive bacteria (P = 0.004) with functions associated with carbohydrate and amino acid metabolism dominated in UB. On the contrary, Gram-negative bacteria with many functions associated with virulence, disease (P = 0.008), defense (P = 5.2 × 10-5), and resistance (P = 0.08) were representative for CB (P values were calculated by Kruskal–Wallis tests; Supplementary Figs. 8–11).
附表2. 每类样品中分箱(Bin)的结果统计
Supplementary Table 2: Summary on binned genomes from the shotgun metagenomics data set.

每列的信息:
- Sample 样品名
- bin No. 分箱编号
- Marker lineage CheckM 评估软件中报导的分类世系
- genomes 基因组编号
- markers 标记基因
- marker sets标记基因集
- 0
- 1
- 2
- 3
- 4
- 5+
- Completeness完整度
- Contamination污染率
- Strain heterogeneity菌株杂合率
- binID 分箱唯一ID
- Count_Function 功能数据
- Fraction_Function 功能比例
- RAST Function RAST功能注释
- Count_OTU OTU数量
- Fraction_OTU OTU比例
- RAST OTU RAST OTU 注释
- RAST subsystem information RAST子系统信息
图4. 基因组概述

重建基因组概述。以平均核苷酸相似度(ANI)结合聚类的高质量基因组,分解为最高分类水平、各自构建的环境起源和各自的复制率(活性)。柱环境色标:蓝色(洁净室设施);红色(重症监护室);深绿色(公共建筑);浅绿色(公共房屋);黄色(私人房屋)
注:洁净空间微生物多样性低,宏基因组更容易获得高质量的Binning。
- Nmeth:DEMIC——使用宏基因组数据预测细菌的生长速率
An overview of reconstructed genomes. High-quality binned genomes clustered by average nucleotide identity (ANI), resolved to highest taxonomic levels, respective built environment origins, and respective replication rates (activity). Color code for column environment: blue (cleanroom facility); red (intensive care unit); dark green (public buildings); light green (public houses); yellow (private houses)
附图8. 革兰氏阳性细菌
Supplementary Figure 8: Gram positive bacteria

基于16S预测样品中革兰氏阳性菌的相对丰度。使用BugBase分析。
Phenotype prediction of Gram positive bacteria based on 16S rRNA gene amplicon analysis.
附图9. 革兰氏阴性细菌
Supplementary Figure 9: Gram negative bacteria

附图10. 致病潜力
Supplementary Figure 10: Potential pathogens

附图11. 潜在胁迫耐受
Supplementary Figure 11: Potential stress tolerance

不足:以上四个图,完全可以拼成一个图的a,b,c,d子图来说明,也避免了图注的重复;图片可以上传emf/wmf的矢量图又小又清晰,而不是模糊的位图
从不同的UB环境中通常可以恢复属于外生细菌Exiguobacterium(V = 0, P = 2.2 × 10-11)和巨球菌Macrococcus(V = 0, P = 1.0)的基因组。节杆菌Arthrobacter(v=465.5,p=2.9×10-15)和两面神菌属Janibacter(v=0,p=0.3)的基因组在公共建筑和公共房屋的类别中更为特异。此外,在私人住宅发现水栖菌属 Enhydrobacter (V = 0, P = 1.0), 考克氏菌属Kocuria (V = 0, P = 8.3 × 10-4), 泛菌属 Pantoea (V = 225, P = 1.2 × 10-9),乳球菌 Lactococcus(V = 9, P = 1.0)和葡萄球菌Staphylococcus(V = 3445, P = 0.01)。明串珠菌Leuconostoc(V = 169, P = 0.9)标志着从私人住宅到重症监护室的转变。最后,基因组分类属于丙酸杆菌Propionibacterium(V = 2697, P = 0.01)、假单胞菌Pseudomonas (V = 133530, P = 2.9 × 10-15)和Stenotrophomonas (V = 97.5, P = 0.07)是所有CB环境的特征(来自Wilcoxon符号秩检验的P值;图4)。在不同构建环境代表性序列物种注释得到了单reads分析数据(附图5,6)和16S rRNA基因扩增子的支持(附图12)。
Genomes assigned to Exiguobacterium (V = 0, P = 2.2 × 10-11) and Macrococcus (V = 0, P = 1.0) were commonly recovered from diverse UB environments. Genomes of Arthrobacter (V = 465.5, P = 2.9 × 10-15) and Janibacter (V = 0, P = 0.3) were more specific for the category of public buildings and public houses. Enhydrobacter (V = 0, P = 1.0), Kocuria (V = 0, P = 8.3 × 10-4), and Pantoea (V = 225, P = 1.2 × 10-9) were found additionally in private houses together with Lactococcus (V = 9, P = 1.0) and Staphylococcus (V = 3445, P = 0.01). Leuconostoc (V = 169, P = 0.9) marked the transition from private houses to ICU. And finally, genomes assigned to Propionibacterium (V = 2697, P = 0.01), Pseudomonas (V = 133530, P = 2.9 × 10-15), and Stenotrophomonas (V = 97.5, P = 0.07) were characteristic to all CB environments (P values from Wilcoxon signed rank tests; Fig. 4). Representative taxonomic assignments for distinct built environments were supported by data of the single-read analysis (Supplementary Figs. 5 and 6) and 16S rRNA gene amplicons (Supplementary Fig. 12).
附图12. 受限不同组特异的16S物种类别
Supplementary Figure 12: Distinctive taxa based on 16S rRNA gene amplicons

主要分为三组,严格控制,中等控制和不受控制的建筑环境。基于LEfSe分析,鉴定不同组特异的Biomarker.
LEfSe analysis (LDA effect size) on 16S rRNA gene amplicons of controlled (gowning area, cleanroom), moderate controlled (ICU) and uncontrolled (public buildings, public and private houses) built environments with the following parameters: per-sample normalization to 1M, factorial Kruskal-Wallis test among classes (alpha = 0.05), pairwise Wilcoxon test between subclasses (alpha = 0.05), threshold for the LDA score (2.0), strategy for multi-class analysis (all-against-all, more strict).
分配给不动杆菌属的基因组(中位完整性94%,中位污染20%)在所有采样建筑环境中高度流行和普遍存在。这使得我们可以对来自不同维护建筑环境的密切相关的细菌物种,在泛基因组水平上的功能特性的变化进行详细的比较。来自私人住宅、重症监护室、洁净室及其更衣室的不动杆菌基因组与所有CDS的24-39%共享核心基因组(核心编码DNA序列与基因组中所有编码DNA序列的比例)。ICU恢复的不动杆菌基因组(如乙酰辅酶A乙酰转移酶FADA或酒精脱氢酶FRMA)中的编码基因与该核心(39%)的重叠度最大,与私人住宅的不动杆菌基因组(2857株特异性CDS,核心基因组的24%)相比,菌株特异性CDS(784)的重叠度更小。对于所有组合的基因组,ICU环境显示其核心基因组(0.2%核心CDS)的最大密度(最相似等级)与所有其他取样构建环境(附表3)相比。不动杆菌的全基因组差异在毒性、疾病和防御功能方面尤为显著。在CB中,分配给这些类别功能的数量几乎是UB的两倍。
注:泛基因组分析,从共有,特有的数量、功能角度分析差异。但Bin不完整且污染过高,结果可信度不高。
Genomes assigned to the genus of Acinetobacter (median completeness 94%, median contamination 20%) were highly prevalent and ubiquitous in all sampled built environments. This has allowed a detailed comparison of closely related bacterial species from different maintained built environments regarding changed functional properties on pan-genome levels. Genomes of Acinetobacter from private houses, the ICU, the cleanroom and its gowning area shared a core genome with 24–39% of all CDS (proportion of core coding DNA sequences to all coding DNA sequences in a genome). Coding genes in the recovered genome of Acinetobacter (e.g., Acetyl-CoA acetyltransferase fadA or alcohol dehydrogenase frmA) from the ICU showed the biggest overlap with this core (39%) and less strain-specific CDS (784) than genomes of Acinetobacter from the private houses (2857 strain-specific CDS, 24% of the core genome). Regarding all binned genomes, the ICU environment showed the greatest density (highest grade of similarity) for its core genome (0.2% core CDS) compared to all other sampled built environments (Supplementary Table 3). Differences in the pan-genome of Acinetobacter were especially striking for functions associated with virulence, disease, and defense. In CB, the number of assigned functions to these categories almost doubled compared to UB.
附表3. 不同环境和物种的泛和核心基因组
Supplementary Table 3: Pan- and Core genome analysis of different built environments and species.

一般来说,与微生物分布图相比,功能特征在所有取样室内空间的分布更加均匀。(附图13—16)。然而,基于SEED注释的LEFSE分析揭示了与革兰氏阳性细菌(革兰氏阳性细胞壁成分、血红素和血红素摄取以及在革兰氏阳性菌中的利用)、脂肪酸代谢(脂肪酸脂质、异戊二烯、磷壁和脂磷壁酸生物合成)、DNA修复系统(DNA修复UvrABC系统、DNA修复细菌Rec FOR通路、转录修复偶联因子)和热激(热激 dnaK基因簇)是UB的显著鉴别特征相关的功能。相反,与革兰氏阴性细菌(革兰氏阴性细胞壁成分)、铁获得(铁色素铁受体、依赖于TONB的铁载体受体和铁载体Pyoverdine)、氧化应激、膜转运和分泌(TON和TOL转运系统、RND外排系统、内膜转运蛋白meB, Type III, IV, VI ESAT分泌系统)、毒力(毒力疾病和防御)和抗性(对抗生素和有毒化合物的抗性、多药抗性外排泵、钴-锌-镉抗性蛋白CzcA)被确定为CB的代表。将所有带注释的SEED功能与RAST服务器引文9,10进行比较,发现UB的氨基酸和碳水化合物代谢功能比例较高(附图17)。相比之下,来自CB的基因组显示出向毒力、疾病和防御等其他功能的转变。尤其是,来自洁净室环境的基因组显示出更为均匀分布的所有功能组,此外,还有许多与压力反应相关的功能。
In general, functional traits were more evenly distributed over all sampled indoor spaces compared to microbial profiles (Supplementary Figs. 13–16). Nevertheless, a detailed LEfSe analysis based on SEED annotations revealed functions associated to Gram-positive bacteria (Gram-positive cell wall components, heme and hemin uptake, and utilization in Gram positives), fatty acid metabolism (fatty acid lipids, isoprenoids, teichoic and lipoteichoic acid biosynthesis), DNA repair systems (DNA repair UvrABC system, DNA repair bacterial Rec FOR pathway, and transcription repair-coupling factor), and heatshock (heatshock dnaK gene cluster) as significant discriminative features of UB. On the contrary, functions associated with Gram-negative bacteria (Gram-negative cell wall components), iron acquisition (ferrichrome iron receptor, TonB-dependent siderophore receptor, and siderophore pyoverdine), oxidative stress, membrane transport and secretion (Ton and Tol transport systems, RND efflux system inner membrane transporter CmeB, Type III, IV, VI ESAT secretion systems), virulence (virulence disease and defense), and resistances (resistance to antibiotics and toxic compounds, multidrug resistance efflux pumps, cobalt zinc cadmium resistance protein CzcA) were identified to be representative for CB. A comparison of all annotated SEED functions with the RAST server9,10 revealed a high proportion of functions associated with amino acid and carbohydrate metabolism for UB (Supplementary Fig. 17). In contrast, genomes from CB indicated a shift towards other functions like virulence, disease, and defense. Especially, genomes from the cleanroom environment showed much more evenly distributed functional capabilities for all functional groups and, additionally, many functions associated with stress response.
附图13. 功能组成柱状图
Supplementary Figure 13: Functional profile (barchart)


使用MEGAN定量单个reads于NCBI NR数据库SEED第一层级的结果。
Single reads BLASTx (rapsearch and diamond) vs. NCBI nr. SEED level 1 (derived from MEGAN, excluding unassigned reads, normalized data set).
附图14. 功能组成放射图
Supplementary Figure 14: Functional profile (radial chart)


使用MEGAN定量单个reads于NCBI NR数据库SEED第一层级的物种级别的功能注释的结果。
Space filling radial chart of SEED annotations on level 1 (species level, excluding unassigned reads, normalized, percentage) assigned (BLASTx NCBInr, diamond and rapsearch) to different built environments (MEGAN).
附图15. 独特的功能
Supplementary Figure 15: Distinctive functions

LEfSe分析(LDA效应大小)对CB(ICU、更衣区、洁净室)和UB(公共建筑、公共和私人住宅)构建环境的宏基因组单个reads功能的注释(根据SEED数据库),参数如下:每个样本标准化到1百分(M),类间因子Kruskal-Wallis检验(alpha=0.05),成对子类间Wilcoxon检验(alpha=0.05),LDA得分阈值(3.0),多类分析策略((all-against-all, more strict)。
LEfSe analysis (LDA effect size) on functions (according to SEED database) of single reads from metagenomes of CB (ICU, gowning area, cleanroom) and UB (public buildings, public and private houses) built environments with the following parameters: per-sample normalization to 1M, factorial Kruskal-Wallis test among classes (alpha = 0.05), pairwise Wilcoxon test between subclasses (alpha = 0.05), threshold for the LDA score (3.0), strategy for multi-class analysis (all-against-all, more strict).
附图16. 物种和功能分别主坐标轴分析
Supplementary Figure 16: Meta-analysis of organisms and functions

从CB和UB环境中获得的宏基因组样本与从植物、城市室内空气和人类微生物群落项目中公开获得的宏基因组样本进行比较分析,通过MG-RAST观察到生物和功能丰度水平。
Comparative analysis of metagenome samples from CB and UB environments with publically available metagenome samples from plants, urban indoor air and the human microbiome project on organism and functional abundance levels visualized through MG-RAST.
附图17. 主要的基因组功能
Supplementary Figure 17: Main genome functions

高质量Bins基于SEED功能注释的相对丰度
Relative proportions of annotated SEED functions with RAST for high quality bins from the metagenomics dataset of all sampled built environments.
抗生素抗性组存在差异
Differences were reflected by the resistome
由于我们对毒力和耐药性相关功能有着明显的了解和兴趣,我们更详细地捕捉了CB和UB的毒力(毒力因子实体)和耐药性(抗生素抗性实体)。CB(19)基因的毒力基因(VFDB)略高于UB(18),在重症监护室中出现的毒力基因比例最高,其次是公共和私人住宅。在高度不受限制的公共建筑环境中,数量最低。因此,CB和UB中染色体编码的细菌毒性可能与其独特的微生物特征有关。但比例差异不显著。(显不显著要用统计说话,不写统计方法和P值就总有显著。而且本研究样本量不大,不容易找到统计显著的差异)
Due to distinct profiles and our interest in functions related to virulence and resistance, we captured the virulome (entity of virulence factors) and resistome (entity of resistances against antibiotics) of CB and UB in greater detail. Slightly more virulence genes (VFDB) were detected for genomes of CB (19) than of UB (18). Highest proportions of virulence genes were present inside the ICU, followed by public and private houses. Lowest counts were visible for the highly unrestricted environment of public buildings. Hence, chromosomally encoded bacterial virulence in CB and UB was likely associated with its distinct microbial profiles. However, differences in proportions were not significant.
病毒组中比较CB与UB的抗性差异更明显**。利用CARD(综合抗药性数据库),对42个选择的优质组合基因组和91个提取的质粒共鉴定出377个不同的抗药性特征**。为了对固有(124)和可移动(186)抗性特征进行详细分析,对检测到的抗性基因进行人工管理(根据参考文献11,去除仅为突变和调节介导的抗性)。CB和UB来自基因组和质粒的抗性有显著差异(变异检验的置换多元分析: n = 37, pseudo-F = 3.8, P = 0.004 和 pseudo-F = 4.0, P = 0.002;图5和附图18)。UB在提取的质粒上比CB表现出更多的可移动性(10比6%)、转座性(36比13%)、复制性(29比10%)和毒力(6比4%)因子或元件。总的来说,基因组和提取的质粒之间的抗性相互连接是非常少见的。只有少数编码不同外排泵(pmrA和acrA)的基因可以在基因和分离出的Exiguobacterium sibiricum、链球菌科(两者均来自UB)和嗜麦芽窄营养单胞菌(位于洁净室设施内)的质粒之间转移(图6),因为它们在相同的环境中被检测到和/或恢复自相似的基因组。然而,它们在抗性中的作用,特别是acrA(这是一个固有的三部分肠杆菌科外排泵的组成部分)仍不清楚。CB显示了与固有抗性有关元件的丰富性,包括外排泵和抗应力决定因素(例如,通过LEfSE分析确定,所有CB环境中的多药外排蛋白mexK和mexB以及过氧化氢酶过氧化物酶激活异烟肼katG)。除了建立环境特异性图谱外,还观察到抗性组的物种特异性模式;例如,嗜麦芽链球菌中的smeA(多药外排)和干酪链球菌基因组中的salA(可能对Lincosamides和链霉素的抗性;图7a、b)。
Compared to the virulome, the resistome showed clearer differences for CB vs UB. Using CARD (Comprehensive Antibiotic Resistance Database), 377 different resistance features could be identified for the 42 selected high-quality binned genomes and 91 extracted plasmids. Detected resistance genes were manually curated (removal of only mutation and regulation-mediated resistances according to ref. 11) for a detailed analysis of intrinsic (124) and mobile (186) resistance features. The resistome of CB and UB as well as resistances from genomes and plasmids differed significantly (Permutational Multivariate Analysis of Variance test: n = 37, pseudo-F = 3.8, P = 0.004 and pseudo-F = 4.0, P = 0.002; Fig. 5 and Supplementary Fig. 18). UB showed more often mobile (10 vs 6%), transposable (36 vs 13%), replication (29 vs 10%) and slightly more virulence (6 vs 4%) factors or elements on their extracted plasmids than CB. Overall, interconnections of the resistome between genomes and extracted plasmids were very rare. Only a few genes encoding diverse efflux pumps (pmrA and acrA) could have been transferred between genomes and extracted plasmids of Exiguobacterium sibiricum, Streptococcaceae (both from UB), and Stenotrophomonas maltophilia (inside the cleanroom facility), respectively (Fig. 6), since they were detected in the same environment and/or recovered from similar genomes. However, the role they might have in resistance, particularly acrA, which forms the part of an intrinsic tripartite Enterobacteriaceae efflux pump, remains obscure. CB showed significantly higher abundance of elements involved in intrinsic resistance, including efflux pumps and stress-resistance determinants (e.g., as identified by LEfSe analysis, the multidrug efflux proteins mexK and mexB, and the catalase peroxidase-activating isoniazid katG in all CB environments). Besides built environment-specific profiles, species-specific patterns of the resistome were also observed; for instance, smeA in S. maltophilia (multidrug efflux) and salA in genomes of Macrococcus caseolyticus (possible resistances against lincosamides and streptogramins; Fig. 7a, b).
图5. 抗性特征的多样性分析

检测到的抗性特性的多样性估计。在CB(受限)和UB(不受限构建环境)以及结合的基因组和质粒中,CARD数据库的不同抗性特征(最高水平,3级)的香农多样性估计有显著差异。数据被标准化(细化),注释数据库使用综合抗生素耐药数据库CARD。(文中展示了Shannon指数综合了丰富度和均匀度,受限环境多样性会显著低,导致抗性相对于本底量相对富集,同时看richness应该更全面)
Diversity estimates of detected resistance features. Significant differences in Shannon diversity estimates of different resistance features (highest levels, level 3) of the CARD database inside CB (confined) and UB (unrestricted built environments) as well as on binned genomes and plasmids. Data were normalized (rarefied). CARD, Comprehensive Antibiotic Resistance Database
附图18. 基因组和质粒的抗性组进行PCoA
Supplementary Figure 18: Resistome of genomes and plasmids

基因于抗性基因注释数量进行主坐标轴分析,对基因组、质粒进行聚类或分类。可见质粒明显聚集。
Distances of binned genomes and plasmids according to detected resistance genes (CARD database).
图6. 基因组和质粒的抗性网络

基因组和质粒的抗性网络。可能转移(边缘连接)的抗性基因(CARD数据库),根据它们在同一构建环境中的组合基因组和质粒中的存在/不存在。边缘加权弹簧嵌入算法在Cytoscape中实现可视化。填充/实心圆代表基因组和空圆代表质粒。抗性基因的丰富度与圆圈大小相关。颜色由各自的建筑环境定义:蓝色(洁净室设施);红色(重症监护室);深绿色(公共建筑);浅绿色(公共房屋);黄色(私人房屋)。数据来源CARD
Resistance network of genomes and plasmids. Potentially transferred (edge-connected) resistance genes (CARD database) according to their presence/absence in binned genomes and plasmids inside the same built environment. Edge-weighted spring-embedded algorithms implemented in Cytoscape were used for visualizations. Filled circles represent genomes and empty circles, plasmids. Most abundant resistance genes were used for labeling and correlated to circle sizes. Colors are defined by respective built environments: blue (cleanroom facility); red (intensive care unit); dark green (public buildings); light green (public houses); yellow (private houses). CARD, Comprehensive Antibiotic Resistance Database
图7. CARD高级分类和药物分类
CARD和药物分类的相对比例。
a. 抗性组高级分析根据CARD注释环境(CB和UB),核苷酸的结构(binned genomes和plasmids),和个个样的分箱基因组(指代单独物种)。
b. 抗性对应的药物组成(CB和UB)。CARD,综合抗生素耐药数据库

Proportion of CARD categories and drug classes. a Higher categories of the resistome according to CARD per environment (CB and UB), nucleotide structure (binned genomes and plasmids), and for individual binned genomes (referring to individual species). b Drug classes and their conferred resistance to them according to CARD per environment (CB and UB), nucleotide structure (binned genomes and plasmids), and for individual binned genomes (referring to individual species). CARD, Comprehensive Antibiotic Resistance Database
CB和UB之间的另一种明显差异是在对不同药物类别的潜在耐药性方面。与UB相比,CBs对氟喹诺酮类(W = 1705, P = 0.4)和三氯生(W = 1666, P = 0.02)的抗性相对丰富。反之,UB对氨基糖苷(W = 1842, P = 0.007)、二氨基嘧啶(W = 1384.5, P = 0.7)和大环内酯类抗生素(W = 1598.5, P = 1.0;P值来自Wilcoxon符号秩检验)。关于它们的位置,编码β-内酰胺、苯尼考和链霉素抗性的基因在结合的基因组中更为常见,而提取的质粒可以调节对氟喹诺酮类、氨基糖苷类和二氨基嘧啶类的更多抗性。同样,Arilatensis节杆菌的基因组对氟喹诺酮类有许多抵抗力,而分配给不动杆菌、假单胞菌和鞘氨醇的基因组对四环素有丰富的抵抗力。嗜麦芽窄食单胞菌对这两种药物都有许多耐药性。与此相反,葡萄球菌科、干酪大球菌和西比灵杆菌的多药耐药更为常见(基于Bin中每类菌的抗性注释组成)。
Further differences between CB and UB were also evident in terms of potentially conferred resistances against distinct drug classes. CBs were relatively enriched by resistances against fluoroquinolones (W = 1705, P = 0.4) and triclosan (W = 1666, P = 0.02) compared to UB. In turn, UBs were more representative of resistances against aminoglycoside (W = 1842, P = 0.007), diaminopyrimidine (W = 1384.5, P = 0.7), and macrolide-based antibiotics (W = 1598.5, P = 1.0; P values from Wilcoxon signed-rank tests). Regarding their location, genes encoding beta-lactam, phenicol, and streptogramin resistance were more common in binned genomes, while extracted plasmids could mediate more resistances against fluoroquinolones, aminoglycosides, and diaminopyrimidines. Likewise, genomes of Arthrobacter arilaitensis showed many resistances against fluoroquinolones, while genomes assigned to Acinetobacter sp., Pseudomonas sp., and Sphingobium were rich in resistances against tetracyclines. Stenotrophomonas maltophilia harbored many resistances to both drug classes. On the contrary, more unspecific multidrug resistances were frequently common for Staphylococcaceae, Macrococcus caseolyticus, and Exiguobacterium sibiricum.
与提取的质粒或不同的构建环境类别(所有质粒中只有20-30%的核心抗性基因)相比,单独结的基因组的核心抗性更为一致(所有基因组中100%的核心抗性基因)。这些数据与固有抗性的概念一致,即一组抗性基因存在于一个特定物种的所有(或大多数)成员中。因此,CB的核心耐药组对氟喹诺酮和氨基香豆素有耐药,而UB则对这些抗生素有耐药,另外对四环素和莫匹罗星也有耐药。
The core resistome of individually binned genomes was much more coherent (100% of core resistance genes in all genomes) than the core resistome of extracted plasmids or the different built environment categories (only 20–30% of core resistance genes in all plasmids). These data agree with the concept of intrinsic resistomes as a set of resistance genes present in all the (or most) members of a given species12. Hence, the core resistome of CB showed resistances against fluoroquinolones and aminocoumarins, while UB contained resistances to these antibiotics and additionally against tetracyclines and mupirocins.
正如已经显示的微生物组的组成,注释的抗性特征也被用来建立预测模型的监督学习方法。如果预测是基于抗性基因(CB与UB: 整理体准确率91%)和微生物组(CB与UB: 整理体准确率92%)的预测是准确的。然而,诸如海拔(R = 0.64, P = 3.3 × 10-3)、温度(R = 0.46, P = 0.09)和微生物丰度(R = 0.46, P = 0.06)等数值环境参数无法轻易预测,仅显示低模型精度。
As already shown for the composition of the microbiome, annotated resistance features were also used to build predictive models by supervised learning methods. Predictions were almost accurate if they were based on resistance genes (CB vs UB: overall accuracy = 91%) instead of microbial profiles (CB vs UB: overall accuracy = 92%). However, numerical environmental parameters like sea levels (R = 0.64, P = 3.3 × 10-3), temperature (R = 0.46, P = 0.09), and microbial abundance (R = 0.46, P = 0.06) could not be predicted easily and showed only low model accuracies.
对抗性基因进行进一步的基因组研究。在大多数情况下,抗生素抗性基因与其他抗性基因共定位,尤其是在从CB环境(主要是多药外输转运系统,如acrA、acrB和bepE)中获得的基因组上。相比之下,来自UB环境的基因组显示,转录调控因子(如cymR和grpE)和转座酶(tnpABC)更常见于带注释的抗性基因附近。尽管转座酶基因在抗性基因附近的频率较高,但不能检测到整合子基因簇。来自CB环境基因组的抗性基因也明显更经常被更高频率的侧翼重复序列所包围(W = 12075, P = 0.02)。在基因组可塑性区域的潜在水平转移基因(HGT)通过同构断裂和CB和UB基因组与MaGee数据库中提供的密切相关基因组之间的成分偏差来识别。在来自CB环境的基因组中检测到更多潜在的HGT特征(可移动基因和tRNA热点)。然而,高比例的血红蛋白在CB中并不显著。
Resistance genes were further investigated in their genomic context (synteny). In most cases, antibiotic resistance genes were co-localized with other resistance genes especially on genomes retrieved from CB environments (mainly multidrug efflux transporter systems e.g., acrA, acrB, and bepE). In contrast, genomes from UB environments showed more often transcriptional regulators (e.g., cymR and grpE) and transposases (tnpABC) in close vicinity to annotated resistance genes. Despite the high frequency of transposase genes in the vicinity of resistance genes, no integron clusters could be detected. Resistance genes of genomes from CB environments were also significantly more often surrounded by a higher frequency of flanking repeats (W = 12075, P = 0.02). Potentially horizontally transferred genes (HGT) in regions of genome plasticity were identified by synteny breaks and the compositional bias between genomes of CB and UB and closely related genomes available in the MaGe database13. More potential HGT features (both mobility genes as well as tRNA hotspots) were detected in genomes from CB environments. However, higher proportions of HGT in CB were not significant.
综上所述,CB表面微生物多样性显著降低(W = 110, P = 1.3 × 10-7)50%,同时抗性显著增加(W = 202.5, P = 0.01)20%,表明在这些环境中,抗性微生物的富集会取代易感微生物(P值来自Wilcoxon符号秩检验)。
In summary, a significant (W = 110, P = 1.3 × 10-7) reduction in microbial diversity on surfaces in CB by 50% was accompanied by a significant (W = 202.5, P = 0.01) increase of resistances by 20%, suggesting an enrichment of resistant microorganisms that displace the susceptible ones in these environments (P values from Wilcoxon signed-rank tests).
讨论
Discussion
我们对深度测序的宏基因组和16S rRNA基因扩增子进行了比较分析,发现建筑表面具有明显的微生物样式具有不同的维持水平。虽然UBs主要是与室外环境和加工食品相关的细菌特征,但CBs显示了大量的序列,主要是与人类相关的细菌、机会性病原体,并且只有很低比例的潜在有益细菌(无潜在病原体;功能比例较低与毒力、防御力和抵抗力有关)。UBs主要是由强大的革兰氏阳性细菌定殖,具有许多功能,以适应微气候、紫外线辐射和营养有效性的波动条件。与此相反,人类相关的革兰氏阴性细菌所选择的持续的中度小气候和强烈的人为影响。定期和严格的清洁程序指导微生物群编码与氧化应激相关的功能,结合膜转运、分泌和凋亡功能,从竞争激烈的营养不良环境中收集营养素,这种环境被描述为微生物的荒地。这些微生物种群的清洁剂和有毒化合物经常暴露在环境中,因为它们的功能增强,能够降解异生物、香叶醇、柠檬烯、蒎烯、萘、双酚、氯环己烷、氯苯、药物代谢,以及总体上更高水平的毒力、疾病、防御和抵抗力。研究的病毒和耐药性强调了人类对建筑环境中微生物的强烈影响。对于需要在清洁、营养不良和微生物控制的CB环境中生存的细菌来说,毒力因子更为丰富。在抗生素耐药性方面,来自CB的细菌倾向于编码更多参与多药外流的基因多样性,而来自UB的细菌则具有更具体的耐药性特征。许多不同抗生素和某些清洁剂的常规应用可能会选择这些微生物控制环境中的广谱耐药特征,并增加对氟喹诺酮类、三氯生或elfamycins的耐药性。与这项研究类似,Lax和同事已经报告了不同AMR在近基因组背景下的共同定位,以及医院相关表面上高比例的多药外排基因(如mexAC)。
Our comparative analysis of deeply sequenced shotgun metagenomes and 16S rRNA gene amplicons revealed a clear microbial pattern on building surfaces characterized by different maintenance levels. While UBs were dominated by bacterial signatures commonly associated with the outdoor environment and processed food, CBs revealed a high abundance of sequences assigned to mainly human-associated bacteria, opportunistic pathogens, and only a low proportion of potentially beneficial bacteria (no potential pathogens; lower proportions of functions associated with virulence, defense, and resistance). UBs were mainly colonized by robust Gram-positive bacteria with many functional capabilities to adapt to fluctuating conditions of the microclimate, UV radiation, and nutrient availability. Opposed to this, the constant moderate microclimate of CB and the strong anthropogenic influence selected for human-associated Gram-negative bacteria. Regular and strict cleaning procedures directed the microbiome to encode for functions associated with oxidative stress in combination with functions for membrane transport, secretion, and apoptosis to gather nutrients from a highly competitive nutrient-poor environment, a condition that was described as a wasteland for microbes14. Regular exposure to cleaning reagents and toxic compounds of these microbial populations were encountered by increased functional capabilities to degrade xenobiotics, geraniol, limonene, pinene, naphthalene, bisphenol, chlorocyclohexane, chlorobenzene, drug metabolism, and an overall higher level of virulence, disease, defense, and resistances. Investigated virulomes and resistomes underlined the strong impact of humans on microbiomes in the built environment. Virulence factors were more abundant for bacteria that need to survive in clean, nutrient-poor, and microbially controlled CB environments. Regarding antibiotic resistance, bacteria from CB tend to encode for a bigger diversity of genes involved in multidrug efflux, while bacteria from UB harbored more specific resistance features. The regular application of many different antibiotics and certain detergents might select such broad-spectrum resistance features in these microbial-controlled environments and also increase resistances against fluoroquinolones, triclosan, or elfamycins. Similar to this study, Lax and coworkers already reported a co-localization of different AMRs in close genomic context and the high proportion of multidrug efflux genes (e.g., mexAC) on hospital-associated surfaces3.
我们的抗性分析不仅包括检测抗性基因存在与否和丰富度,还包括它们在各自的基因组草图中的水平以及它们与已知病原体水平转移的潜力。除此综合分析外,本研究还面临一些局限性,如来自CB环境的样本量较低、集中于一种样本类型——地板样本,以及缺乏特定抗生素的元数据。特别是与其他研究相比,ICU在取样时缺乏元数据。这种低样本量是由于ICU和洁净室设施的受限建筑环境设置以及这些CB环境中的低生物量导致的。因此,后续分析的代表性是有限的,也限制了我们将微生物、毒力或抗性成分与环境变量关联和解释的尝试,如Lax等人2017年的研究所示。因此,所述结果的一般有效性和影响需要进一步研究和确认。
Our resistome analysis covered not only the presence and abundance of detected resistance genes, but also their context in respective draft genomes as well as their potential to be horizontally transferable with known pathogens2,5,15. Besides this comprehensive analysis, the present study faces some limitations, such as the low sample size from CB environments, its focus on one sample type (floor samples), and the lack of metadata on specific administered antibiotics especially in the ICU at the time of sampling in contrast to other studies3,4. This low sample size was a consequence of the restricted access to the confined built environment setting of the ICU and the cleanroom facility as well as the low amount of biomass in these CB environments. Hence, the representativeness of the subsequent analysis is limited and also constrained our attempts to correlate and interpret microbial, virulence, or resistance compositions with environmental variables, as was shown in the study of Lax et al. in 20173. Therefore, the general validity and impact of the presented results require additional confirmation by further studies.
然而,我们的研究试图从三个方面证实CB和UB之间的抗性组差异。首先,抗药性特征的多样性增加与潜在病原体的数量呈正相关。其次,我们在基因组可塑性、转座酶、侧翼重复和整合子簇中定位潜在的水平转移基因,以及质粒的抗药性以覆盖可移动的遗传元件。最后,我们对完整的微生物细胞进行了分类分析,并确定了复制水平,以强调代谢活性微生物群内的抗性。面对这种差异化的分析,CB的抗性更为多样,可能具有移动性,并且与潜在病原体的接触更为频繁,但通常活性更低,因此在取样时更难操纵。在微生物多样性总体下降的情况下,CB的这些方面表明了不利的人为影响。许多研究强调了微生物多样性在稳定微生物群落和抵御病原体入侵方面的作用。因此,功能多样性和组成多样性可以被认为是生态系统稳定性的一个非特定但普遍的标志。当前的研究连同先前发表的工作,强调了微生物多样性的丧失与耐药性的增加有关,这表明这些种群可能承载更多抗生素抗性有机体。可以想象,生物多样性的恢复可能会减少抗生素耐药性。
Nevertheless, our study tried to substantiate the assessment of observed differences of the resistome between CB and UB by three aspects. First of all, the increased diversity of resistant features in CB was positively correlated with the number of potential pathogens. Secondly, we targeted potentially horizontally transferred genes in regions of genome plasticity, transposases, flanking repeats, and integron clusters as well as the resistome of plasmids to cover mobile genetic elements. And finally, we differentiated our analysis for intact microbial cells and determined the level of replication to emphasize the resistome inside metabolically active microbiota. Facing this differentiated analysis, the resistome of CB was more diverse, potentially mobile, and in increased contact to potential pathogens, but often less active and therefore harder to manipulate at the time of sampling. These aspects of CB in the presence of an overall decreased microbial diversity indicate an adverse anthropogenic influence. Many studies emphasize the role of microbial diversity to stabilize microbial communities and to act as a protection shield against the invasion of pathogens16,17,18. Hence, functional and compositional diversity can be considered as an unspecific, but universal, marker of ecosystem stability19. The current study, together with the previously published work7, highlights that the loss of microbial diversity correlates with an increase of resistances, indicating that these populations might be burdened by antibiotic-resistant organisms. It is conceivable that the restoration of biodiversity may allow a decrease of antibiotic resistance.
然而,虽然洁净室是强制性的要求几乎没有微生物,但是医院、私人或公共建筑中的其他区域不需要(或可以)没有微生物。此外,出于卫生目清洁并不意味着有必要使用抗菌产品,这会对耐药性产生不利的选择压力。考虑到人类干预减少微生物负荷可能导致微生物多样性下降,这与微生物组中抗生素耐药性的增加有关,人类暴露在几乎无菌的环境中应限于手术室或洁净室的特定操作过程。生命活动的建筑环境中应该有更高的微生物多样性,增加微生物多样性的一个简单方法是通过定期的窗户通风来增加空气与室外环境的交换。或者,正如我们之前提议的,至少在封闭区域附近引进绿色植物。另一个步骤是对室内和卫生保健环境进行主动控制。生物控制已经为其他应用建立了方法;室内的一项研究表明,通过将芽孢杆菌孢子应用于医疗保健设施,取得了很有前景的结果。
However, while it is mandatory for cleanrooms to be almost free of microorganisms, other areas in hospitals or in private or public buildings do not need (or can) to be absent of microorganisms. Furthermore, cleaning for hygiene purposes does not imply the necessity to apply antimicrobial products that would propel adverse selection pressure on the resistome. Given that human interventions for reducing microbial load may cause a decline in microbial diversity, which is associated with the increase of antibiotic resistance in the microbiome, human exposure to almost sterile environments should be limited to operating rooms or particular industrial processes in cleanrooms. All other areas of life in the built environment could be enriched by a higher microbial diversity. One simple solution to increase microbial diversity is to increase the exchange of air with the outdoor environment by regular window ventilation. Or, as we proposed before, to introduce green plants, at least in close vicinity to confined areas20,21,22. Another step would be the active manipulation or ‘biocontrol’ of indoor and health-care environments23,24. Biocontrol is established already for other applications;19 a first study indoors showed promising results through the application of Bacillus spores in a health-care setting25.
建筑物是人们生活、共享微生物的主要环境,许多与人类活动有关的疾病可能有其起源。此外,微生物分布受微生物维持和建筑限制的影响。然而,由于有效的免疫发展可能依赖于微生物暴露,因此在建成环境中对许多微生物的非选择性去除和杀灭可能会对健康产生不利影响。特别是,在CB环境中检测到的这种广谱选择机制容易损害微生物群,这将导致生物多样性丧失,并可能对世代产生累积效应。因此,建筑环境的限制应限于上述规定的区域和特殊要求。对于所有其他的建筑环境,建筑材料可能是多种多样的,以允许更高的微生物多样性。表面维护(例如清洁)可以是多样化的,并且杀生物剂的应用可以局限于热点位置和不同的时间段。最后,还需要仔细考虑抗菌剂在建筑物中的总体使用。
Buildings are the main environment in which people spend their lives, share microbes, and where many diseases associated with anthropogenic activities may have their origin26. Moreover, microbial profiles are affected by microbial maintenance and building confinement27. However, an unselective removal and killing of many microbes in the built environment could have adverse health effects, since potent immune development may rely on microbial exposure23,28,29,30. In particular, such broad-spectrum selection mechanisms detected in CB environments are prone to damage the microbiome, which would lead to a loss of biodiversity and possibly to an accumulating effect over generations17. Hence, the confinement of a built environment should be limited to defined areas and special demands as indicated above. For all other built environments, building materials could be diverse to allow a higher microbial diversity31. Surface maintenance, such as cleaning, could be diversified, and the application of biocidal detergents can be limited to hot-spot locations and distinct timeframes. Also, in the end, the overall use of antimicrobials in buildings needs to be carefully considered.
健康建筑内存在高度多样、稳定和设计的有益微生物组可能导致未来我们处于抗性组中的风险降低。由于微生物组的总体遗传力远低于人类通过其在建筑中的行为获得微生物组的10倍,因此我们不应因为抗菌抗性替换而失去数百万人,而是应该重新考虑我们在建筑环境中的行为。
The presence of highly diverse, stable, and beneficially designed microbiomes inside healthy buildings could result in lower exposures to resistances in the future. Since the overall heritability of microbiomes is much lower (up to 10-fold) than human beings acquire microbiomes by their behavior in, e.g. buildings32, we are not condemned to lose millions of people due to antimicrobial resistances—instead it is time to reconsider our behavior in the built environment.
方法
Methods
环境参数和研究设计
Environmental parameters and study design
在一年中的同一季节(春季),对各种不同的室内环境进行了微生物控制、维护和接触。所有这些室内环境都具有表1中总结的不同环境参数,这些参数被怀疑有助于微生物组的组成和功能。关于研究设计和潜在环境影响和差异的更多细节,可以在补充方法(附图19-21和附表4、5)中找到。研究了两种不同清洁度的室内环境地板:UB无限制建筑(公共建筑、公共和私人住宅)和CB受限建筑(重症监护室和无尘室设施)。如下文所述,通过16S rRNA基因扩增子和鸟枪法宏基因组测序对种群结构和整个宏基因组组成进行研究。
A variety of indoor environments different in their levels of microbial control, maintenance, and access were sampled during the same season of the year (spring). All these indoor environments featured different environmental parameters summarized in Table 1, which were suspected to contribute to the composition and function of their microbiome. More details about the study design and potential environmental influences and differences can be found in the Supplementary Methods (Supplementary Figs. 19–21 and Supplementary Tables 4, 5). Two types of floors of indoor environments with different cleanliness levels were investigated: UB, unrestricted buildings (public buildings, public and private houses) and CB, confined buildings (intensive care unit and cleanroom facility). The structure of the population and the whole metagenomic composition were investigated through 16S rRNA gene amplicon and shotgun metagenomic sequencing as described below.
附图19. 非限制的建筑环境(UB)
Supplementary Figure 19: Unrestricted buildings (UB)

来自公共建筑、公共和私人房间的地图,地点为德国Grossenaspe野生动物园
Sampling map of public buildings (L) and public § and private houses (F) in a wildlife park in Grossenaspe, Germany (Figure was adapted from https://www.wildpark-eekholt.de/besucherinformationen_lageplan.htm).
附图20. 限制的建筑环境-ICU
Supplementary Figure 20: Controlled built environment (CB) - Intensive Care Unit

奥地利ICU取样示意图
Sampling map of the intensive care unit (ICU) at the state hospital in Graz, Austria (Figure was adapted from 1).
附图21. 限制的建筑环境-洁净室
Supplementary Figure 21: Controlled built environment (CB) - Cleanroom facility

附表4. 用于采样建筑环境中各种表面和使用的清洁和消毒试剂
Supplementary Table 4: A list of cleaning and disinfection reagents applied for various surfaces and purposes in the sampled built environments.

附表5. 包括奥地利格拉茨国立医院ICU中消毒剂种类和使用时间
Supplementary Table 5: A list of cleaning and disinfection reagents including the exposure time applied for certain cases in the ICU at the state hospital in Graz, Austria

表1. 环境参数
Table 1 Environmental parameters

意大利都灵Thales Alenia空间洁净室的取样地图
Sampling map of the Thales Alenia space cleanroom facility in Turin, Italy (Figure was adapted from 2).
取样过程
Sampling procedures
大规模收集地板样品(根据每个房间的大小确定),以获得大生物量(即使是从清洁室等低生物量环境中)。此外,地板样本显示其使用者具有较高的诊断能力,以及较高比例的抗微生物抗性。对于这种方法,无菌(高压灭菌)和无DNA(干热处理)Alpha Wipes® (TX1009; VWR International GmbH, Vienna, Austria)被安装在生物罩下的大拭子(Swiffer® Sweeper® Floor Mop Starter Kit; Procter & Gamble Austria GmbH, Vienna, Austria)由无菌且无DNA的叶片分隔成几层。必要时,用喷雾瓶直接在表面喷洒PCR应水擦拭拭子。所有仪器都经几步进行化学消毒,(all-purpose cleaner, Denkmit, dm-drogerie markt GmbH + Co. KG, Karlsruhe, Germany; 70% (w/v) ethanol, Carl Roth GmbH & Co. KG, Karlsruhe, Germany and Bacillol® plus, Bode Chemie GmbH, Hamburg, Germany)。剩余的DNA用氯漂白剂(DNA away; Molecular Bio Products, Inc., San Diego, CA, USA)和紫外线(254 and 366 nm; Kurt Migge GmbH, Heidelberg, Germany)变性。以重复的方式收集样品,始终从每个室内环境(尤其是根据其ISO分类的洁净室)的清洁区域开始,以尽量减少污染物的转移。由同一个人进行采样,以确保一致的扫掠模式(水平、垂直和对角扫掠运动)以及对颗粒和微生物的一致吸收。在每次取样事件后12小时内,将样品储存在蓝色冰上并在实验室进行处理。在先前的一项研究中,已经获得并处理了重症监护室的样本,但现在已纳入比较分析。
Large-scale floor samples (defined by the size of each room) were collected to obtain high amounts of biomass (even from low-biomass environments like cleanrooms). In addition, floor samples were shown to have high diagnostic capacities of its occupants4 as well as high proportions of antimicrobial resistances3. For this approach, sterile (autoclaved) and DNA-free (dry heat treatment) Alpha Wipes® (TX1009; VWR International GmbH, Vienna, Austria) were mounted in several layers separated by sterile, DNA-free foliage on a big swab (Swiffer® Sweeper® Floor Mop Starter Kit; Procter & Gamble Austria GmbH, Vienna, Austria) under a biohood. If necessary, wipes were remoistened by spraying polymerase chain reaction-grade water directly on the surface with a spray bottle. All instruments were chemically sterilized in several steps (all-purpose cleaner, Denkmit, dm-drogerie markt GmbH + Co. KG, Karlsruhe, Germany; 70% (w/v) ethanol, Carl Roth GmbH & Co. KG, Karlsruhe, Germany and Bacillol® plus, Bode Chemie GmbH, Hamburg, Germany). The remaining DNA was denatured with chlorine bleach (DNA away; Molecular Bio Products, Inc., San Diego, CA, USA) and UV light (254 and 366 nm; Kurt Migge GmbH, Heidelberg, Germany). Samples were collected in a repetitive way, always starting from cleaner areas in each indoor environment (especially in cleanrooms according to their ISO classifications) to minimize the transfer of contaminants. Sampling was executed by the same person to guarantee a consistent sweeping pattern (horizontal, vertical, and diagonal sweeping motions) as well as a consistent uptake of particles and microbes. Samples were stored on blue ice and processed at the laboratory within 12 h after each sampling event. Samples from the ICU were already obtained and processed in a previous study33, but are now included for comparative analysis.
取样处理和DNA提取
Sample processing, PMA treatment, and DNA extraction
在DNA提取之前,对样本进行处理、浓缩和用PMA处理(更多细节在补充方法中提供)。对来自高生物量环境的样品进行PMA处理,作为对所用试剂和取样设备中潜在DNA污染物的额外控制(PMA可以与裸露的DNA结合,用于去除遗迹DNA)。此外,PMA处理还可以作为评估完整微生物细胞比例的方法,并验证数据集中对活微生物细胞得出的结论。使用SDS的DNA提取方法适用于低生物量样本;但是对于生物量较高的样品,需要根据制造商的说明使用GeneClean®Turbo Kit(MP Biomedicals,海德堡,德国)进行额外处理。
Samples were processed, concentrated, and treated with PMA prior to DNA extraction (more details are provided in Supplementary methods). PMA treatment of samples from high-biomass environments was performed as an additional control for potential DNA contaminants in used reagents and on sampling equipment. In addition, PMA treatment served as a proxy to evaluate the proportions of intact microbial cells and validate drawn conclusions on viable microbial cells in the dataset. The DNA-extraction method with the xanthogenate-SDS (XS) buffer was suitable for low-biomass environments; however for samples with higher biomass, an additional treatment with the Geneclean® Turbo Kit (MP Biomedicals, Heidelberg, Germany) according to manufacturer’s instructions was necessary.
定量测量
Quantitative measures
采用定量聚合酶链反应对大多数样品的细菌丰度进行了研究。更多细节见补充方法。
Bacterial abundance was investigated for most samples by quantitative polymerase chain reaction. Further details are specified in Supplementary methods.
宏基因组测序
Shotgun metagenomics
将所有公共建筑、公共住宅、私人住宅、重症监护室、洁净室和更衣区样品的总提取DNA汇总(平均DNA量约10μg,浓度为149 ng/μL)。经质量控制后,通过对DNA进行机器打断和末端修复,制备出9个鸟枪法文库。插入片段大小为300 bp。在Eurofins Genomics GmbH (Ebersberg, Germany) 使用Illumina Hiseq 2500测序仪在快速运行模式下使用PE150模式下进行测序。
Total extracted DNA of all the samples was pooled into the categories of public buildings, public houses, private houses, ICU, and cleanroom and gowning area with a mean DNA amount of ~ 10 µg and a mean DNA concentration of 149 ng/µl. After quality control, nine shotgun libraries were prepared by fragmentation and end repair of DNA with insert sizes of ~ 300 bp. Sequencing was performed at Eurofins Genomics GmbH (Ebersberg, Germany) using an Illumina HiSeq 2500 instrument with 2 × 150 bp paired ends in the rapid run mode.
16S rRNA基因扩增与测序
16S rRNA gene amplicons and sequencing
用带有条形码的引物对 515F – 806R(引物序列见附表6)产生针对16S rRNA基因的扩增子。更多细节可在补充方法中找到。
Amplicons targeting the 16S rRNA gene were generated with the barcoded primer pair 515f–806r34,35 (primer sequences are listed in Supplementary Table 6). Further details can be found in Supplementary methods.
附表6. 本研究中使用的引物
Supplementary Table 6: Complete list of all primers used in the study.

实验对照
Controls
除实际样品外,在每个实验步骤对阴性对照进行处理。PMA处理作为对所用试剂、设备和总体观察中低生物量环境中自由仍可扩增背景DNA的额外质量控制。提取对照和空白对照(背景环境的样本不与地板表面接触)并行处理。在下面描述的生物信息学分析过程中,从标准化数据集中减去这些控制样本的序列。
Negative controls were processed at each experimental step besides the actual samples. PMA treatment served as an additional quality control for free still-amplifiable background DNA in used reagents, equipment, and overall observations of low-biomass environments. Extraction controls and field blanks (samples of the background environment without any contact with the floor surface) were processed in parallel. Sequences of these control samples were subtracted from the normalized dataset during the bioinformatics analysis as described below.
生信分析
Bioinformatics
宏基因组:原始序列进行质量控制后,采用phred值 > 35 且最小长度为50进行过滤,同时移除测序接头(附表7. 样本信息统计表)
附表7. 宏基因组数据集统计
Supplementary Table 7: Summary on all quality reads of the shotgun metagenomics dataset.

包括样本名,总测序量,序列长度,GC含量,双端序列量和平均质量
全部的分析以基因组为中心,重点关注组装的数据,如重叠群,骨架和分箱。然而,基因中心的分析基于单个reads,因此整个分析中存在组装的人造序列(artifacts)。
Shotgun metagenomics: after the quality control of raw reads, sequencing adapters were removed from sequences and quality filtered according to phred score ( > q35) as well as length filtered (min. 50 bp) by trimming from the 3’ prime site (Supplementary Table 7). The whole analysis was conducted in a genome-centric approach focusing on assembly-based data (contigs, scaffolds, and bins). However, gene-centric analysis based on single reads served as a quality control for assembly-related artifacts throughout the analysis.
采用Blastx算法(软件是diamond,比blastx快上百倍)比对序列至自定义的标记数据库(eggNOG4.0里所有的COGs/NOGs,现在数据库版本为5.0,包括99%的古菌、细菌和真核),也同时比对了NCBI非冗余数据库(版本211,2015年12月)。所有的注释采有MEGAN软件实现。对于基因组中心的方法,Ray Meta 组装高质量的序列,K-mer的长度为31。组装结果采用如下参数过滤:最小长度1500,最小覆盖度5,read长度150。组装结果的摘要见附表8。
附表8. 宏基因组组装的重叠群(contig)和骨架(scaffolds)统计
Supplementary Table 8: Summary of all contigs and scaffolds after assembly of the shotgun metagenomics dataset.

共4个表,分别为contig 和 scaffold大于100和500-nt筛选下的评诂结果,主要包括数量、总长度、平均长度取整、N50、中位数和最大值。
These single reads were assigned using BLASTx search algorithm with default settings36 against a custom-marker database (all COGs/NOGs in eggNOG 4.037, which can be found in 99% of all archaea, bacteria, and eukaryota) as well as against the NCBI non-redundant database (release 211.0 of December 2015). Annotations of all single reads were determined and analyzed with MEGAN (MEtaGenome ANalyzer)38. For the genome-centric approach, quality sequences were assembled with Ray Meta and a k-mer length of 3139. Assemblies were filtered according to the following parameters: minimum length 1500, minimum coverage 5, and read length 150. A summary of all filtered assemblies is provided in Supplementary Table 8.
过滤后的重叠群采用AMPHORA2进行物种分类,并采用以上的提到的标记基因数据库。然后采用Krona绘图,并将每个重叠群的覆盖率作为丰度。这些重叠群进一步采用CONCOCT和MaxBin进行分箱(现在流程MetaWRAP),Bins的质量采用CheckM进行评估,详见附表2。
Afterwards, the filtered contigs were taxonomically classified with AMPHORA240 using the database of markers described above. For the visualization in Krona charts, the coverage ratios of respective contigs were considered to show relative abundances. Contigs were further binned through a genome-centric approach with CONCOCT41 and MaxBin42. Binning quality of contigs was validated with CheckM43 (Supplementary Table 2).
基因组草图的完整度在75–85%,且污染率在2–25%范围内,被认为适合下游分析。每个Bin的重叠群采用AMPHORA重注释,与RAST和MaGe比对公共可用的基因组来挖掘生态相关功能的子系统和关注泛基因组(核心基因组和可变基因组,MicroScope基因/蛋白家族(MICFAMs) 参数:80%按氨基酸相似度,80%比对覆盖度),病毒组(balstp比对至VFDB数据库,采用MaGe下60%的相似度,仅选择最佳匹配),抗性组(CARD同源和变异,版本v1.1.2,官方配套软件RGI v3.1.145)。
Draft genomes in the range of 75–85% completeness and 2–25% contamination were considered to be suitable for downstream analysis. Contigs of each bin were re-annotated with AMPHORA2 and compared with publicly available genomes using RAST9 and MaGe13 to reveal ecologically relevant functional subsystems with special focus on pan-genomes (core genome and variable genome; MicroScope gene/protein families (MICFAMs) parameters: 80% amino acid identity, 80% alignment coverage), virulomes (running BLASTp on organism proteins against MicroScope, the virulence factor database VFDB44 accessed in MaGe13 with 60% identity and considering only best hits), and resistomes (CARD homologs and variants, v.1.1.2, RGI v.3.1.145).
进一步的共线性分析详见补充方法。蛋白的功能注释采用比较基因组IMG的GO、KEGG和SEED分类。采用** Recycler 提取质粒**,并采用KEGG、UniRef90和CARD注释(详见补充方法)。
Further details about the synteny analysis can be found in Supplementary methods. Predicted functional classifications of protein-coding genes were analyzed by annotation and comparative genomics in IMG46 with GO terms (Gene Ontology Consortium, 2000), KEGG (Kyoto Encyclopedia of Genes and Genomes)47, and SEED classifications9. Plasmids were extracted with Recycler48 and annotated with KEGG, uniref90, and CARD (see Supplementary methods for more details).
分箱基因组与dRep比较,采用iRep计算复制率。分类算法的表型调研(PICA)用于预测Bin的表型。更详细的生信工具详见附表9。基于16S rRNA序列的生信分析群体结构详见补充方法。
附表9. 生信分析软件参数
Supplementary Table 9: Settings for selected bioinformatic tools.

主要软件:Trimmomatic-0.32用于数据质量过滤;MEGAN 5用于序列可视化;Ray Meta-2.3.1组装;CONCOCT-0.4.0和MaxBin1.4.2分箱;Amphora2物种注释;dRep-0.5.7基因组比较;iRep-1.1基因组复制速率计算。
Binned genomes were compared with dRep49 and replication rates were calculated with iRep50. Phenotype Investigation with Classification Algorithms (PICA) was used to predict the phenotypes of binned genomes (phendb.org)51. More details on the settings of used bioinformatic tools can be found in Supplementary Table 9. Bioinformatic analyses of the population structure based on 16S rRNA sequences are described in detail in Supplementary methods.
统计信息
Statistical information
统计分析采用QIIME 1.9.1, QIIME2 2017.10和2018.11中的R脚本,或直接在R中使用vegan包。统计检验,包括比较分类,距离 ,距离矩阵,核心微生物、核心微生物组和核心功能,分类信息汇总,共发生样式,元数据相关,生物环境检验详见附表10-12,以及多元线性回归模型。
Statistical analyses were conducted in QIIME 1.9.1 and QIIME 2 versions 2017.10 and 2018.1152 (calling respective R scripts) or directly in R53 using the vegan package. Statistical tests included a comparison of categories, distances, distance matrices, core microbiomes and core functions, taxa summaries, co-occurrence patterns, correlations of metadata, a bioenv test (Supplementary Tables 10–12), and multivariate linear regression models.
附表10. 16S数据上应用的统计方法
Supplementary Table 10: Summary of applied statistics on the 16S rRNA gene amplicon dataset.

采用MRPP、adonis和ANOSIM统计区间距离显著性,以及对差异的解析率;t检验用于组间比较,BEST寻找最佳环境因子;使用了(unweighted)unifrac和Bray-Curtis两类距离。
附表11. 对PICRUSt结果统计的方法和显著性
Supplementary Table 11: Summary of applied statistics on predicted functions from the 16S rRNA gene amplicon dataset with PICRUSt.
基本同附表10
附表12. 16S数据的样本统计信息
Supplementary Table 12: Read statistics of the 16S rRNA gene amplicon dataset.

包括样本数,OTUs数量,总数据量;样本数据量,以及基本摘要统计。
对于无参检验,使用多响应转换方法,如adonis分析相似度,转换多元回归方差分析,Kruskal–Wallis, Kolmogorow–Smirnow, Wilcoxon秩和检验(Mann–Whitney U test),Spearman秩相关,箱线图比较距离,Mantel相关图和Mantel检验,统计显著性基于999次置换检验。箱线图中的距离比较采用双尾t检验,Bonferroni校P值。
For nonparametric tests like multi-response permutation procedures, adonis, analysis of similarities, permutational multivariate analysis of variance, Kruskal–Wallis, Kolmogorow–Smirnow, Wilcoxon signed-rank test (Mann–Whitney U test), Spearman rank correlations, distance-comparison box plots, Mantel correlograms, and Mantel tests, statistical significance was determined through 999 permutations. Distance-comparison box plots were calculated using a two-sided Student’s two-sample t test. All resulting P values were Bonferroni corrected.
PCoA图基于有权重的unifrac距离,NMDS计算基于Bray-Curtis距离。展示在NMDS中的环境因子采用bioenv函数计算,基于欧式距离,并添加每个样品组的置信椭圆。LEfSe统计生物标志物,和microPITA挑选感兴趣的分类丰度。所有分析采用默认参数。采用microbiome包分析特征的差异丰度。样本元数据预测采用QIIME2中的分类和回归模型。
PCoA plots were based on weighted unifrac metrics. NMDS was calculated from a Bray–Curtis distance matrix. Vectors of environmental variables shown in NMDS were calculated with the bioenv function based on Euclidean distances in R as were calculated ellipses per sample group. LEfSe54 and microbiome: picking interesting taxonomic abundance analysis (microPITA) (http://huttenhower.sph.harvard.edu/micropita) were performed on Galaxy modules provided by the Medical University of Graz (https://galaxy.medunigraz.at/). Both tools were executed with default settings using an all-against-all strategy for the multi-class analysis for 16S rRNA gene amplicon datasets as well as CARD annotations and a one-against-all strategy for the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States-predicted functions throughout the LEfSe analysis. Differential abundance of features was calculated with analysis of composition of microbiomes55. Sample metadata was predicted with random forest classification and regression models in QIIME 256.
猜你喜欢
- 10000+: 菌群分析
宝宝与猫狗 提DNA发Nature 实验分析谁对结果影响大 Cell微生物专刊 肠道指挥大脑 - 系列教程:微生物组入门 Biostar 微生物组 宏基因组
- 专业技能:生信宝典 学术图表 高分文章 不可或缺的人
- 一文读懂:宏基因组 寄生虫益处 进化树
- 必备技能:提问 搜索 Endnote
- 文献阅读 热心肠 SemanticScholar Geenmedical
- 扩增子分析:图表解读 分析流程 统计绘图
- 16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
- 在线工具:16S预测培养基 生信绘图
- 科研经验:云笔记 云协作 公众号
- 编程模板: Shell R Perl
- 生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习扩增子、宏基因组科研思路和分析实战,关注“宏基因组”


点击阅读原文,跳转最新文章目录阅读
https://mp.weixin.qq.com/s/5jQspEvH5_4Xmart22gjMA