南海水产研究所姜敬哲团队、香港城市大学孙燕妮团队、广东药科大学原丽红合作开发的可用于病毒快速分类生信工具
使用PhaGCN2对病毒基因组片段分类
Virus classification for viral genomic fragments using PhaGCN2

文章链接:https://www.researchsquare.com/article/rs-1658089/v1
软件链接:https://github.com/KennthShang/PhaGCN2.0
第一作者:Jing-Zhe Jiang(姜敬哲), Wen-Guang Yuan(袁文广), and Jiayu Shang(商家煜)
通讯作者:Jing-Zhe Jiang (姜敬哲),Yanni Sun(孙燕妮), Li-Hong Yuan(原丽红)
合作作者:Ying-Hui Shi(史莹慧), Li-Ling Yang(杨李玲), Min Liu(刘敏), Peng Zhu(朱鹏),Tao Jin(金桃),
主要单位:
南海水产研究所 (Key Laboratory of South China Sea Fishery Resources Exploitation & Utilization, Ministry of Agriculture and Rural Affairs, South China Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Guangzhou 510300, Guangdong, China)
中国药科大学 (Guangdong Province Key Laboratory for Biotechnology Drug Candidates, School of Biosciences and Biopharmaceutics, Guangdong Pharmaceutical University, Guangzhou 510006, Guangdong, China)
香港城市大学(Department of Electrical Engineering, City University of Hong Kong, Hong Kong (SAR), China)
天津农学院(Tianjin Agricultural University, Tianjin 300384, China)
广东美格基因(Guangdong Magigene Biotechnology Co., Ltd, Guangzhou 510000, Guangdong, China)
摘要
Abstract
背景:病毒是生物群系中最普遍和最多样化的实体。由于新发现病毒的快速增长,我们迫切需要新的工具来对新的病毒进行准确且全面的病毒分类。
结果:本研究中,我们提出了PhaGCN2,它可以在科水平上快速对病毒序列进行分类,并支持网络图来可视化结果文件。我们使用几个广泛使用的指标将PhaGCN2与最先进的病毒分类工具(如vConTACT2、CAT和VPF-Class)进行比较。结果表明,PhaGCN2大大提高了病毒分类的precision和recall。基于PhaGCN2预测结果,我们将GOV2.0(全球海洋病毒数据库)数据库中的可分类病毒序列数量提高了4倍,并且分类了90%以上的GPD数据库(人体肠道病毒数据库)。PhaGCN2使国际病毒分类委员会(International Committee on Taxonomy of Viruses)的高通量和自动扩充数据库成为可能。
引言
Introduction
作为地球上最丰富的生物实体,病毒几乎可以寄生所有的生命体。它们通过影响宿主死亡、代谢、生理和进化来影响海洋生物地球化学循环。生物测序技术最近被用于从不依赖培养的环境样本中探索病毒圈的多样性。病毒基因组数据库的迅速扩张导致国际病毒分类委员会(ICTV)提出了一项共识声明,建议从“传统”分类标准转向以基因组为中心的分类。目前,病毒分类主要依靠病毒学家手工分类和定义,对数百万个病毒基因组序列进行分类太慢。例如,尽管IMG/VR中有数百万种病毒序列,但ICTV2021年报告(以下简称ICTV2021)中分类的病毒只有约10,550种。因此,迫切需要一种病毒分类方法,能够快速和准确地对这些新的病毒基因组序列进行分类,并将计算分类与ictv批准的分类单元对齐。
在我们之前的工作中,我们提出了一个基于图卷积网络的半监督机器学习模型,—— PhaGCN。PhaGCN中有两个主要组件:CNN编码器和GCN分类器。首先,CNN编码器将不同长度的contigs编码为256维的嵌入向量。每个向量代表从DNA序列中捕获的相关图案。其次,建立一个knowledge graph,将RefSeq数据库中的已知噬菌体与测试噬菌体连接起来。图中的每个节点代表一个噬菌体,噬菌体之间的边代表基于序列和蛋白质组成的相似性。我们使用CNN编码器输出的嵌入向量作为节点特征,并应用蛋白质簇和蛋白质相似性来定义边的特征。最后,在knowledge graph上应用GCN,利用已知噬菌体和测试噬菌体进行训练。但是目前的版本只能对Caudovirales(有尾噬菌体目)下的病毒进行分类。更重要的是,ICTV会根据研究的进展经常调整其分类标准,如删除旧科,增加新科,将成员从一个科转移到另一个科。参考文献的不断变化和新病毒的出现,阻碍了自动预测的准确性和敏感性。另外,大多数基于学习的模型必须指定标签集(例如,科标签),这将不能容纳来自新科的病毒。因此,需要一种能识别新科的方法来支持病毒自动分类。
在这里,我们提出了PhaGCN2,一个可以通过升级数据库,使分类与ICTV批准的分类标准保持一致的病毒分类工具。PhaGCN2可以在科水平上预测病毒序列的分类,准确识别ICTV中尚未定义的新病毒科成员。我们使用几个广泛使用的指标将PhaGCN2与最先进的病毒分类工具(如vConTACT2、CAT和VPF-Class)进行比较。实验结果表明,该方法优于现有方法.
结果
Result
PhaGCN2的主要改进
Improvements of PhaGCN2
PhaGCN2与以前的版本相比有三个主要的改进,包括(1)使用prodigal在整个病毒领域下构建参考数据库(2)使用网络图来识别离群点,(3)将离群节点分配给family_like。(2)和(3)中的改进使PhaGCN2能够自动识别新的科,从而消除了常用监督学习模型中对固定标签集的限制。这些改进使得PhaGCN2比原始版本获得了更准确的预测,precision(公式(1))从73.19%提高到83.91%,recall(公式(2))从87.92%提高到89.30%,F1-score(公式(3))从79.88%提高到86.52%(附表1)。详细的描述可以在下面的章节中找到。
 (1)
(1)
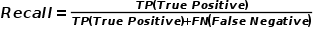 (2)
(2)
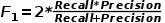 (3)
(3)
蛋白数据库的构建. PhaGCN蛋白质数据库是通过从美国生物技术信息中心(NCBI)手工下载蛋白质序列来构建的。使用旧数据库有两个潜在的缺点。首先,蛋白质的数量受到RefSeq蛋白质数据库更新的限制。其次,用户需要逐个序列地将蛋白质映射到原始基因组,这是一项繁琐且容易出错的工作。为了建立更快捷、更方便用户的数据库构建管道,我们应用Prodigal基于最新的ICTV2021数据库进行了蛋白翻译(最新的ICTV2021包含10550个病毒)。利用DOV (Dataset of Oyster Virome)中的8760个病毒序列(length>8000bp)作为测试序列,将使用Prodigal构建的数据库的PhaGCN2与原始PhaGCN数据库进行比较。结果显示98.46%的预测结果是一致的,说明使用Prodigal建立蛋白质数据库是可靠的(表S2)。现在,用户可以通过训练PhaGCN2中的病毒分类数据库的功能,将分类与ICTV批准的分类进行对齐。
网络图可视化. 与vConTACT2类似,PhaGCN2也可以输出病毒簇集群网络。这可以我们对不同病毒科和病毒科成员之间的关系有了直观的了解。除了将科与科之间的关系可视化,我们还使用网络拓扑来识别可能的新家庭,这些新科由与ICTV节点弱连接的子图组成。首先,我们识别离群点——没有连接到ICTV中的任何病毒的测试病毒(节点)(图S1A,红点)。通常这些异常值来自新的科,但由于监督学习算法的设计限制,它们被分配到family_like(图S1B,绿点)。
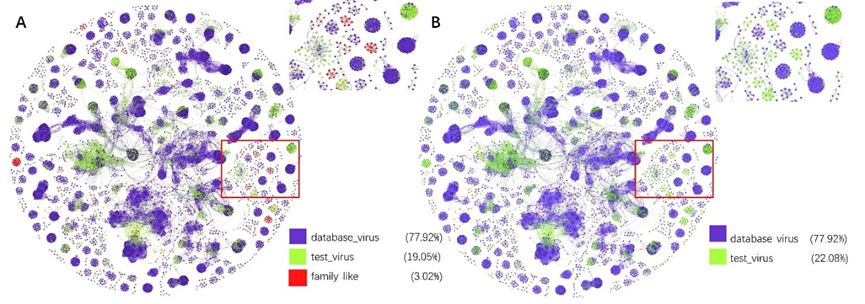
图S1,引入“family_like“前后的网络图对比
图A为代码改动前,图B为代码改动后,图中右上角为红框放大部分,20_database_virus是数据库的点,21_test_virus和family_like是测试数据。
Family-like的预测.为了支持新family_like的自动识别,我们将这些异常值赋值为family_like(可能属于与参考科相近的另一个科)。例如,如果一个节点被预测为Lipothrixviridae_like,这意味着该节点与Lipothrixviridae很接近,但不建议将其归入同一科。为了验证family - like离群值预测的可行性,我们利用ICTV2020病毒构建蛋白质数据库,并利用ICTV2021新添加的病毒(包括2636个病毒参考基因组)作为测试数据。
在新增的2636个病毒中,有339个属于ICTV2020中没有定义的科,因此我们的训练数据中不存在它们的标签。PhaGCN2总共分配了204个病毒作为family_like病毒。在这些序列中,167个测试序列是ICTV2021真正的新科成员或ICTV2020训练时未包含的科成员。因此,family_like标签的precision为81.86%(167/204),recall为49.26%(167/339)。167个真正的类科标签中,有153病毒在ICTV2021中被定义为Genomoviridae,这个科是新增的科,而在ICTV2020中被定义为Geminiviridae(与Genomoviridae同为Geplafuvirales)。改良后的PhaGCN2则将它们直接判定为Geminiviridae_like,提示该病毒很可能属于跟Geminiviridae很相似的一个科。根据ICTV2021,其他37个测试序列被错误地标注为family_like,因为它们是ICTV2020列表中的家庭成员。例如,在ICTV2021中,一些病毒属于Myoviridae,但被PhaGCN预测为Drexlerviridae(与Myoviridae同属Caudovirales)。PhaGCN2将它们识别为Drexlerviridae_like。根据ICTV2021标准,它们属于Myoviridae,但属于该科的一个新属,与ICTV2020中的Myoviridae成员没有交集。事实上,37个测试序列中的大部分在ICTV2021中被归为一个新属。
PhaGCN2与当前最先进的工具的对比
Comparison with the state-of-the-art tools
为了对PhaGCN2进行综合评价,我们使用6个广泛使用的指标:precision(公式(1))、recall(公式(2))、F1-score(公式(3))、consistency (式(4))计算速度和峰值内存,将PhaGCN2与vConTACT2、CAT和VPF-Class进行比较。
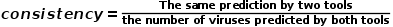 (4)
(4)
Precision、recall、F1-score.我们用PhaGCN2数据库所收录的9604条病毒基因组序列(即ICTV2021序列)作为PhaGCN2,vConTACT2,CAT的测试数据,将得到的结果与ICTV2021的分类进行对比。对比结果如表1。
Table 1.Comparison of PhaGCN2 with the state-of-the-art virus classification tools
Tools | PhaGCN2 | vConTACT2 | CAT | VPF-Class |
Test Data | 96041 ICTV2021 (including 3189 RNA virus2) | |||
True Positive | 8379 | 1616 | 6928 | 3840 |
False Positive | 260 | 773 | 825 | 3683 |
False Negative | 965 | 4026 | 1852 | 2080 |
Precision | 96.99% | 67.64% | 89.36% | 51.04% |
Recall | 89.67% | 28.64% | 78.91% | 64.86% |
F1-score | 93.19% | 40.24% | 83.81% | 57.13% |
1以ICTV2021中超过1700 bp的病毒基因组作为测试数据,对所有软件进行评估。
2由于vConTACT2只为DNA病毒分类而设计,因此将RNA病毒基因组排除在评估之外。
Consistency.我们使用PhaGCN2和CAT对GOV 2.0数据集进行了测试,并将预测结果分别与GOV2.0论文中vConTACT2预测结果进行了对比(图1)。比较发现:①vConTACT2与PhaGCN2共有的预测结果是20430条,结果相同的序列是16958条,一致性为82.97% (16958/20430);②vConTACT2与CAT共有的预测结果是10779条,结果相同的序列是5893条序列,一致性为54.67% (5893/10779); ③CAT与PhaGCN2共有预测结果的序列是13996条,结果相同的序列是5694条序列,一致性为40.68% (5694/13996); ④三者共有的预测结果序列是5441条,结果相同的序列是3205条序列,一致性为58.90% (3205/5441)。结果表明,vConTACT2与PhaGCN2由于采用的分类方法相似,因此预测结果一致性最高,而二者与CAT的结果一致性都不高。
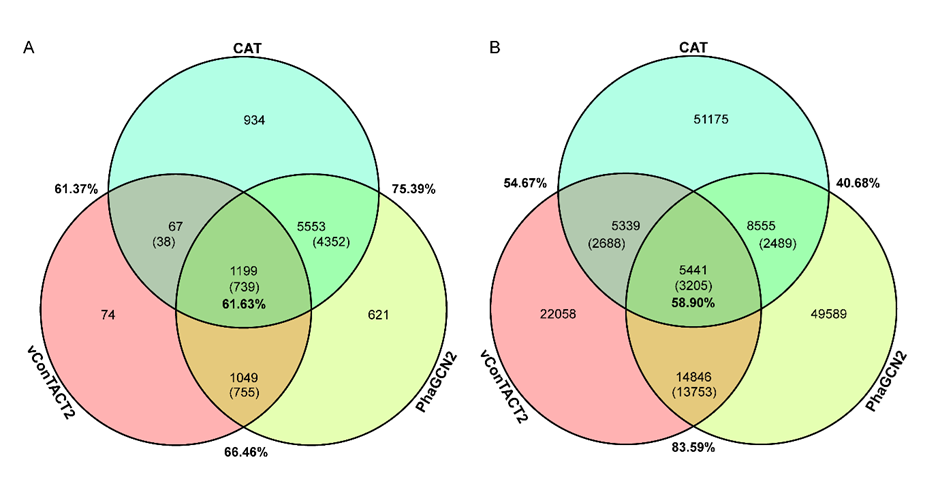
图1:三种病毒分类软件结果一致性的维恩图。
A:测试数据是9604条ICTV2021序列。B:测试数据是482,522 条GOV2.0的序列。不带括号的数为有预测结果的序列数,带括号的数为对应工具有相同预测的序列数,百分比为两个工具之间或三个工具之间的一致性
计算速度和内存峰值.此外,我们记录了这三种工具的运行时间和峰值内存。我们从GPD中随机选择1000、5000和10000个序列进行检测(图2)。
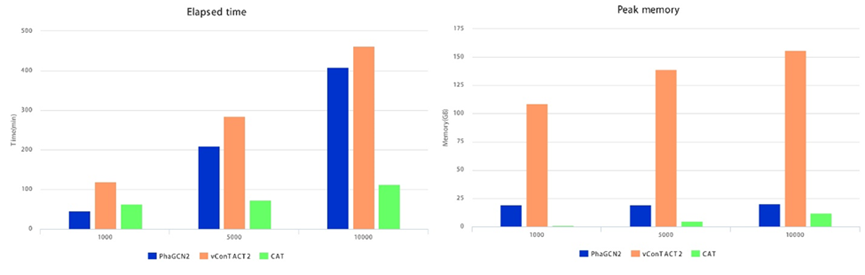
图2:PhaGCN2与vContact2和CAT在运算速度和内存使用上的比较
1000、5000、10000代表测试的基因组序列数量。
对漏检序列的分析
Analysis of the sequences without predictions
在以ICTV2020做训练数据库,以ICTV2021新增的2634条序列作为测试数据的实验中,其中1492条序列结果是阳性的,1142条序列结果是阴性的。我们针对这1142条阴性结果进一步的分析(附表7),其中有992条是ICTV2021新增的科,是本身就不应该被预测到的。剩余150条序列中,有80条序列是已知科下面的新属。我们推测,由于这些科中不同属差异性太大,导致这些新的属没有被预测到;另外有49条序列被漏检,虽然它们不是新属,但由于2020数据中该属下的样本量太少(少于8条),所以没有被PhaGCN2训练。剩余21条序列中我们暂时无法确定原因,然而相对于总共2634条测试序列来说,我们认为这个数字是可以接受的。
进一步,我们以ICTV2021新增序列作为query,以ICTV2020的训练数据作为subject,用diamond blastx进行了对比,比较了阳性和阴性结果组间的Identity、score。结果如图3所示,两组间的Identity有显著的差别,其中Identity在54.8,的新病毒序列会有很大概率被PhaGCN2预测到;而Identity低于37.4%高度变异序列获得阳性结果的概率则很低。
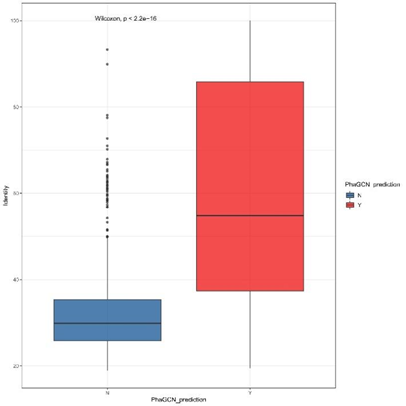
图S2检测序列(阳性结果与阴性结果)与参考基因组之间的蛋白序列一致性比较。N:阴性结果;Y:阳性结果。
PhaGCN2对属水平预测的可能性
Possibility of genus-level prediction
和vConTACT2一样,PhaGCN2也可以绘制网络图。我们使用1700个人类肠道微生物组DNA病毒的基因组作为测试数据,并根据PhaGCN2的结果绘制网络图。由于空间的限制,我们只显示数据库中最大的10个家族的结果(图3A)。显然,同一科的病毒节点紧密地聚集在一起。为了在属水平上可视化簇,我们选取了病毒种最多的Siphoviridae。同样,在图3B中,Siphoviridae的前16个属成员用不同的颜色显示出来。我们可以看到一些属,如Pahexavirus, Skunavirus,和ceduvirus,在自己内部聚集。然而,一些属(如Triavirus、Phietavirus、Bioseptimavirus、Dubowvirus和Peeveelvirus)混合在一起(图3B)。这表明它们的差异不足以使PhaGCN2预测它们是不同的属。
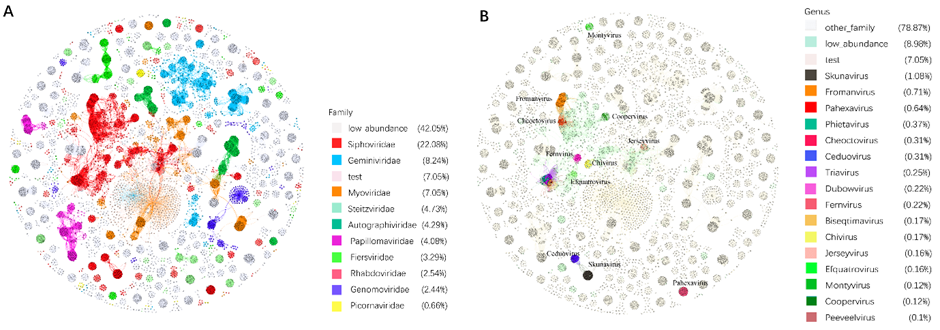
图3,PhaGCN2网络图在科和属水平上的聚类效果展示
AB两图的拓扑结构完全相同,test为MGV测试数据。A图中丰度为前9的科用不同颜色标识,没有着色的low_abundance代表其它低丰度的科;B图中专门显示Siphoviridae科内的不同属,高丰度属(成员数量≧10)用不同颜色标识,low_abundance用浅绿色标识,代表Siphoviridae中其它低丰度的属,other_family代表非Siphoviridae科。
PhaGCN2对公共数据库的挖掘
Investigation of public data using PhaGCN2
GPD和GOV2.0代表了两个完全不同的病毒生境。在本节中,我们使用PhaGCN2对GPD和GOV2.0数据库进行分类。去除不合格序列后,它们分别剩下142333(142809)和328173(482522)。如图4所示,GPD和GOV2.0的总体召回率分别为91.9%和40.8%。GOV2.0中未知病毒的比例较高,远远超过GPD,说明海洋中的病毒还没有得到充分的探索,还有很大一部分仍在冰山下。仅从分类类别来看(没有unknown), Siphoviridae和Myoviridae占GPD的54.5%,Siphoviridae_like和Myoviridae_like占GPD的31.1%。与GPD相比,在GOV2.0中Siphoviridae和Myoviridae占28.9%,Siphoviridae_like和Myoviridae_like占40.4%。如果包括Caudovirales下的其他科,如Podoviridae和Herelleviridae,人类肠道中99.16%的噬菌体为Caudovirales,而海洋中为94.8%。这意味着在目水平上,GPD和GOV2.0以Caudovirales为主,而在科水平上GPD和GOV2.0差异较大。具体结果见表S9和表S5。
我们进一步应用PhaGCN2对2202个合格的RNA病毒基因组进行了分类,这些基因组来自无脊椎动物和脊椎动物病毒的研究。有1094个序列被预测,只有6个病毒基因组被预测为非rna病毒。排在前3位的是马氏病毒科、双裂病毒科和结节病毒科,占总数的18.7%。然而,有高达52.5%的病毒不能被分类到一个已知的病毒家族,这表明我们对RNA病毒圈的了解仍然非常有限。详细结果如表S10和图S3所示。
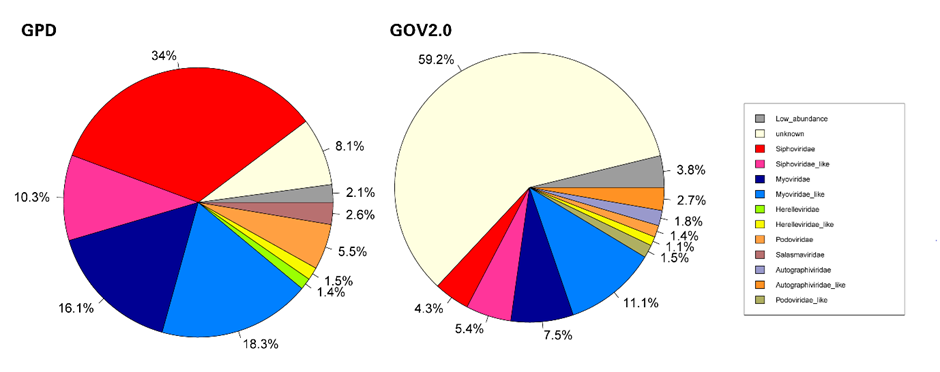
图5,基于PhaGCN2的GPD和GOV2数据中的病毒科水平组成结果比较
该图为GPD和GOV2数据库中使用PhaGCN2进行分类后根据各个科占据的百分比画出来的饼状图,其中“-“后面的数字代表序列条数,low_abundance表示数量较少(低于总个数的0.5%)的科的总和,unknown代表未能预测的数量,其他分别代表各个科。GPD和GOV2.0总测试点分别为142809和482522
进一步,针对GOV2在科水平的分类及来源站位信息,我们还绘制了Myoviridae和Siphoviridae在不同站点和海水深度的分布丰度图(图6),详细经纬度及其含量数据见附表11。由图6可见,Myoviridae表现出非常明显的纬度和深度效应,越接近赤道地区、表层水域,Myoviridae占比越高,而Siphoviridae则是俩极区域显然要超过赤道地区。这说明不同科的病毒可能在漫长的进化过程中对同环境进化出了独特的适应性。
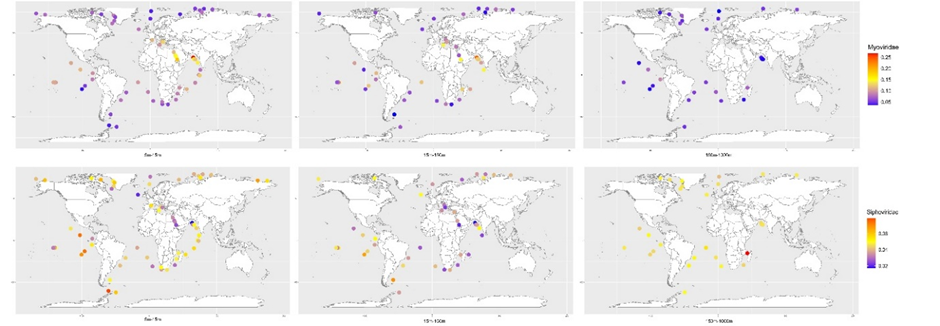
图6:GOV2的Myoviridae和Siphoviridae病毒类群随纬度和海水深度的不同分布模式
地图中颜色深度代表Myoviridae和Siphoviridae占该取样点中总病毒种类百分比含量的大小,颜色代表含量的高低。从左至右的采样深度依次为5m-15m,15m-150m,150m-1000m
讨论
Discussion
vConTACT2是当前被广泛认可的病毒分类工具,它结合了ClusterONE、分层聚类和马尔可夫聚类算法(MCL)生成的蛋白质聚类。该方法的优点是可以准确预测具有多个orf和频繁重组的大型DNA噬菌体的基因组分类。然而,对于含有较少蛋白质簇的噬菌体,其性能会下降。PhaGCN将基于蛋白质聚类的特征集成到一个更强大的基于图卷积网络的机器学习模型中,从而以更少的计算资源获得更高的精度。但是,PhaGCN仅限于噬菌体,这限制了它对病毒的全面分类。PhaGCN2通过扩展学习模型和参考数据库来消除这个限制。PhaGCN2可以应用于所有类型的病毒宏基因组数据,自动产生DNA和RNA病毒的科水平的分类。此外,它还可以根据网络拓扑结构提出新的病毒族。基于比对的分类方法如CAT,BLAST仅依赖比对结果,基于多数投票方法来推断出物种的分类。尽管CAT是识别已知病毒的第二精确的工具(表1),但基于比对的工具在分类新病毒或高度分化的病毒方面并没有优化。
PhaGCN2被设计和训练用于在家族水平上进行预测。虽然该方法可以推广到属水平预测,但许多属的成员数量较少,不足以训练一个广义学习模型。另一个情况是,在当前的ICTV标准下,有些属类过于相似,无法有效区分(图3B)。然而,随着ICTV参考数据集的不断增长和ICTV对属的调整,病毒实现属水平的预测将更加可行。
与其他基于学习的模型一样,PhaGCN2的性能也依赖于训练数据的质量。由于测序的偏重,目前的训练数据没有系统地覆盖不同的分类群。虽然PhaGCN利用网络拓扑结构来预测新家族,但其对新家族的预测能力是有限的。Identity小于37.4%的未知病毒序列的检出率通常很低(图S2)。利用PhaGCN2进行迭代预测是增强新病毒科分类的一种可能策略。首先,我们可以使用PhaGCN2对所有病毒基因组数据(如IMG/VR)进行预测。然后,我们可以将新预测的Family_like成员加入到训练数据中,增加PhaGCN2识别更多新家族成员的能力。迭代训练和搜索可能会提高PhaGCN2对新家族的检测能力。我们将在今后的工作中对此进行研究。
然而,对于那些没有相似度或相似度很低的“暗物质”序列,进行从头病毒分类可能是一项不可能的任务。首先,我们无法评估预测的准确性。其次,没有任何同源物,很难描述它们基因组的结构或功能。无论确定了多少个序列,它们仍然是“暗物质”。
最后,由于PhaGCN2不能预测输入序列是属于病毒还是宿主细胞,我们强烈建议使用病毒序列作为PhaGCN2的输入。换句话说,在应用PhaGCN2之前,应先使用病毒识别工具(如DIAMOND、Virsorter2等)去除非病毒序列。
参考文献
Kunkun Fan, Manuel Delgado-Baquerizo, Xisheng Guo, Daozhong Wang, Yong-guan Zhu & Haiyan Chu. (2020). Biodiversity of key-stone phylotypes determines crop production in a 4-decade fertilization experiment. The ISME Journal, doi: https://doi.org/10.1038/s41396-020-00796-8
Gelderblom, H.R. (1996) Structure and Classification of Viruses. Medical Microbiology.
Suttle, C.A. (2007) Marine viruses βÄî major players in the global ecosystem. Nature Reviews Microbiology, 5, 801-812.
Geoghegan, J.L. and Holmes, E.C. (2017) Predicting virus emergence amid evolutionary noise. Open Biol, 7.
Asokan, G.V. and Kasimanickam, R.K. (2013) Emerging Infectious Diseases, Antimicrobial Resistance and Millennium Development Goals: Resolving the Challenges through One Health. Cent Asian J Glob Health, 2, 76.
Grant, W.B. (2008) Hypothesis--ultraviolet-B irradiance and vitamin D reduce the risk of viral infections and thus their sequelae, including autoimmune diseases and some cancers.
Gregory, A.C., Zayed, A.A., Conceicao-Neto, N., Temperton, B., Bolduc, B., Alberti, A., Ardyna, M., Arkhipova, K., Carmichael, M., Cruaud, C. et al. (2019) Marine DNA Viral Macro- and Microdiversity from Pole to Pole. Cell, 177, 1109-1123 e1114.
Camarillo-Guerrero, L.F., Almeida, A., Rangel-Pineros, G., Finn, R.D. and Lawley, T.D. (2021) Massive expansion of human gut bacteriophage diversity. Cell, 184, 1098-1109 e1099.
Roux, S., Paez-Espino, D., Chen, I.A., Palaniappan, K., Ratner, A., Chu, K., Reddy, T.B.K., Nayfach, S., Schulz, F., Call, L. et al. (2021) IMG/VR v3: an integrated ecological and evolutionary framework for interrogating genomes of uncultivated viruses. Nucleic Acids Res, 49, D764-D775.
Simmonds, P., Adams, M.J., Benko, M., Breitbart, M., Brister, J.R., Carstens, E.B., Davison, A.J., Delwart, E., Gorbalenya, A.E., Harrach, B. et al. (2017) Consensus statement: Virus taxonomy in the age of metagenomics. Nat Rev Microbiol, 15, 161-168.
Dutilh, B.E., Varsani, A., Tong, Y., Simmonds, P., Sabanadzovic, S., Rubino, L., Roux, S., Munoz, A.R., Lood, C., Lefkowitz, E.J. et al. (2021) Perspective on taxonomic classification of uncultivated viruses. Curr Opin Virol, 51, 207-215.
Shang, J., Jiang, J. and Sun, Y. (2021) Bacteriophage classification for assembled contigs using graph convolutional network. Bioinformatics, 37, i25-i33.
Hyatt, D., Chen, G.L., Locascio, P.F., Land, M.L., Larimer, F.W. and Hauser, L.J. (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 11, 119.
Jing-Zhe, J., Yi-Fei, F., Hong-Ying, W., Ying-Xiang, G., Li-Ling, Y., Tao, J., Mang, S., Shao-Kun, S., Meng, W., Tuo, Y. et al. (2021) Dataset of Oyster Virome and the Remarkable Virus Diversity in Filter-Feeding Oysters. Research Square.
Nayfach, S., Páez-Espino, D., Call, L., Low, S.J., Sberro, H., Ivanova, N.N., Proal, A.D., Fischbach, M.A., Bhatt, A.S., Hugenholtz, P. et al. (2021) Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome. Nature Microbiology, 6, 960-970.
Shi, M., Lin, X.-D., Tian, J.-H., Chen, L.-J., Chen, X., Li, C.-X., Qin, X.-C., Li, J., Cao, J.-P., Eden, J.-S. et al. (2016) Redefining the invertebrate RNA virosphere. Nature, 540, 539-543.
Shi, M., Lin, X.-D., Chen, X., Tian, J.-H., Chen, L.-J., Li, K., Wang, W., Eden, J.-S., Shen, J.-J., Liu, L. et al. (2018) The evolutionary history of vertebrate RNA viruses. Nature, 556, 197-202.
Nepusz, T., Yu, H. and Paccanaro, A. (2012) Detecting overlapping protein complexes in protein-protein interaction networks. Nat Methods, 9, 471-472.
Lima-Mendez, G., Van Helden, J., Toussaint, A. and Leplae, R. (2008) Reticulate representation of evolutionary and functional relationships between phage genomes. Mol Biol Evol, 25, 762-777.
Bin Jang, H., Bolduc, B., Zablocki, O., Kuhn, J.H., Roux, S., Adriaenssens, E.M., Brister, J.R., Kropinski, A.M., Krupovic, M., Lavigne, R. et al. (2019) Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat Biotechnol, 37, 632-639.
Altschul, S.F., Gish, W., Miller, W., Myers, E.W. and Lipman, D.J. (1990) Basic local alignment search tool.
Benjamin, B., Chao, X. and H, H.D. (2015) Fast and sensitive protein alignment using DIAMOND. nature methods 12.
Guo, J., Bolduc, B., Zayed, A.A., Varsani, A., Dominguez-Huerta, G., Delmont, T.O., Pratama, A.A., Gazitua, M.C., Vik, D., Sullivan, M.B. et al. (2021) VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome, 9, 37.
von Meijenfeldt, F.A.B., Arkhipova, K., Cambuy, D.D., Coutinho, F.H. and Dutilh, B.E. (2019) Robust taxonomic classification of uncharted microbial sequences and bins with CAT and BAT. Genome Biol, 20, 217.
Pons, J.C., Paez-Espino, D., Riera, G., Ivanova, N., Kyrpides, N.C. and Llabres, M. (2021) VPF-Class: Taxonomic assignment and host prediction of uncultivated viruses based on viral protein families. Bioinformatics.
Shang, J. and Sun, Y.J.M. (2020) CHEER: hierarCHical taxonomic classification for viral mEtagEnomic data via deep leaRning.
M, B., S, H. and M, J. (2009) Gephi: an open source software for exploring and manipulating networks. International AAAI Conference on Weblogs and Social Media.
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读