在后基因组时代,蛋白质组学在生物医学研究中发挥着重要作用。近日,Nature子刊《Laboratory Investigation》发表了一篇高通量蛋白组的mini-review,概述了高通量蛋白质组学技术、统计和算法的进展。

蛋白质组学作为蛋白质组实验和数据分析的结合,从整体上分析了蛋白质的组成、结构、表达、修饰状态以及蛋白质之间的相互作用和联系。它为基因组学和转录组学提供补充信息。它对于生成复杂的、相互关联的通路、网络和分子系统的图谱也是至关重要的,这些通路、网络和分子系统直接控制着主要的生命活动,如细胞的增殖、分化、衰老和凋亡。随着过去十年实验技术的大幅提高,蛋白质组学方法已经从传统的免疫组织化学(IHC)染色、western blot和酶联免疫吸附试验(ELISA),发展到高通量的方法,如组织微阵列(TMA)、蛋白质通路阵列和质谱分析。这些高通量蛋白质组学技术不仅减少了分析时间,还提高了蛋白质组覆盖的准确性和深度。随着生物信息学和现代多分析 "组学 "技术的发展,蛋白质组学在揭示疾病的分子机制,发现新的生物标志物方面有着巨大的前景,并可作为特定的诊断检测、预后预测和治疗目标,进一步加强个性化医疗。
高通量蛋白质组学技术
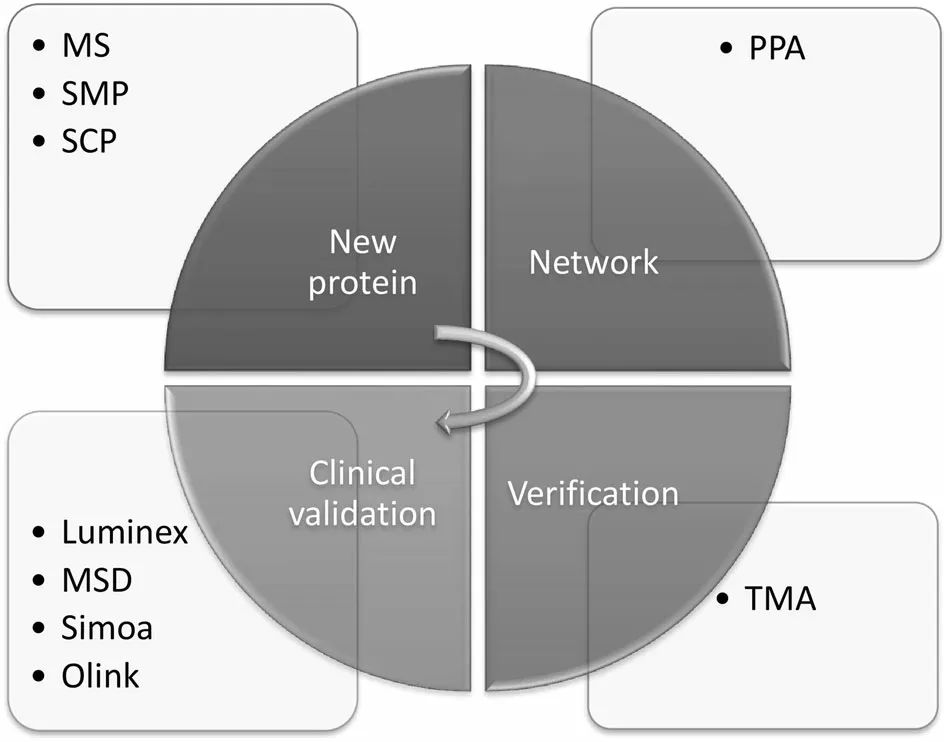
蛋白质组学“从实验到临床”
质谱分析(Mass spectrometry)
质谱(MS)已经发展成为识别蛋白质及其异构体的最基本和最流行的工具之一,并通过直接的片段或负责其形成的特定蛋白质分解活动对翻译后修饰进行量化。MS最重要的作用是发现和检测一个完整的蛋白质或复合肽或代用肽的子集。MS可以与多种分离和预分馏技术相结合,以识别目标蛋白/肽,提高识别的准确性和产量。例如2D-PAGE基于电荷和分子量,而液相色谱(LC)基于极性、电荷和蛋白质分子量。尽管2D-PAGE传统上被用作蛋白质组学研究的标准程序,但基于凝胶的技术往往是劳动密集型和耗时的,因此不适合于高通量蛋白质组学。相比之下,液相色谱或高效液相色谱(HPLC)可以从复杂的混合物中连续分离数千种蛋白质,并可与质谱结合成为LC-MS,以提高通量。其中,反相液相色谱法(RPLC)是最常用的基于LC的分离平台。
根据不同的处理策略,基于质谱的方法可分为自上而下、自下而上和鸟枪法。在自上而下的蛋白质组学方法中,全长的蛋白质可以随后在质谱内被分割,并记录片段的质量,直接被送去进行质谱分析。相比之下,在自下而上的蛋白质组学技术中,蛋白质被酶解或化学消化成肽输入到质谱设备中。此外,鸟枪蛋白质组学是自下而上蛋白质组学的一个特殊案例,复杂混合物(如血清、尿液和细胞裂解液)中的整个蛋白质被切割成肽,然后进行多维HPLC-MS,其目的是像基因组 "鸟枪 "测序一样产生蛋白质混合物的全局概况。另一方面,在自下而上的策略中,不一定需要在MS之前分离多肽。然后,通过依赖数据的发现引擎,对MS数据进行匹配,以确定蛋白质序列数据库中的目标蛋白质及其相关修饰,这可分为肽段评分、蛋白质评分,最后是蛋白质推断。
蛋白通路芯片(Protein pathway array)
蛋白质通路阵列(PPA)是一个基于凝胶的高通量平台,采用抗体混合物来检测蛋白质样品中的抗原,这些样品可以从活检或组织中提取。在这种方法中,可以对肿瘤组织进行显微切割,以最大限度地提高来自肿瘤组织而不是周围良性组织的蛋白质的比例。然后,将抗体-抗原反应的免疫荧光信号转换为数值数据,作为蛋白质表达的数值。在数据归一化和适当的统计学建模后,可以探索和训练生物标志物和蛋白质组网络。PPA已被应用于许多疾病,如原发性血小板增多症和甲状腺乳头状癌。其高通量的蛋白质谱系以稳健的定量方式提供了比传统方法更多的优势。
高通量组织芯片(Next generation tissue microarrays)
随着过去十年技术的进步和高通量需求的增加,TMA逐渐开始在研究和临床领域广泛使用。TMA包含了数百个不同病例的福尔马林固定石蜡包埋(FFPE)或冷冻块的许多小的代表性组织,以阵列方式组装在一张组织学玻片上,因此可以同时对多个样本进行大规模的基于抗体的分子分析。因此,它是确认和验证由PPA或MS蛋白质组学方法产生的新生物标志物的一个实用和有价值的工具。因此,它经常被用于独立的队列,并确定目标蛋白在细胞膜、细胞质或细胞核中的位置。由于在过去的一年里,具有多种智能显微镜的数字病理学得到了快速发展,最近创建了一种新的TMA方法,即高通量组织微阵列(ngTMAs)。它允许将注释直接放在数字玻片上,以获得更高的准确性。ngTMAs的两个主要优点是其时间效率和高通量而不影响质量。由于其改进的灵敏度和快速、大规模的检测能力,ngTMAs已成为提高临床和转化研究中使用的TMA质量的有力工具。
基于磁珠或适体的多重检测(Multiplex bead- or aptamer-based assays)
近年来,基于Luminex珠阵列系统越来越多地被用于蛋白质分析应用中。它使蛋白质组学生物标志物panel的检测可靠、快速,并能应对各种临床实践中的动态变化。另一个广泛使用的平台是Meso-scale Discovery (MSD),对于人类细胞因子谱,MSD测定比Luminex测定更敏感,但特异性较低。Simoa®是广泛使用的bead-based的多重检测之一。它涵盖6个疾病领域,可定制,截至2022年5月,包括109个肿瘤学、26个神经学、19个免疫学、13个心脏病学和45个传染病检测。基于适体的SOMAscan®分析可在单个检测中评估1000至9000种抗原的表达,并具有广泛的动态范围(8个数量级)、极高的灵敏度(检测下限,40fm)和高精度(中位变异系数=5%)。
邻位延伸技术(Proximity extension assay (Olink)
PEA作为邻近依赖性连接分析之一,基于彼此具有轻微亲和力的寡核苷酸连接抗体对。当这些寡核苷酸连接的抗体接近时,与抗体连接的两个独特的寡核苷酸将被DNA聚合酶延伸,并在随后以指数方式扩增。Olink检测法已经成功应用于多个临床领域,包括2019年冠状病毒疾病(COVID-19)、创伤性脑损伤和肾脏疾病。它可以在极少量的样品(如几微升)中同时对3000多种蛋白质进行定量。
基于纳米孔的单分子蛋白质组学(Nanopore based single-molecule proteomics
纳米孔已越来越多地用于DNA或RNA测序,并尝试用于蛋白质组学。它在蛋白质组学的早期应用是在分枝杆菌肽的测序中。基于纳米孔的单分子蛋白质组学的主要挑战是检测翻译后修饰(PTM)的效率低和缺乏足够的灵敏度。
统计和算法
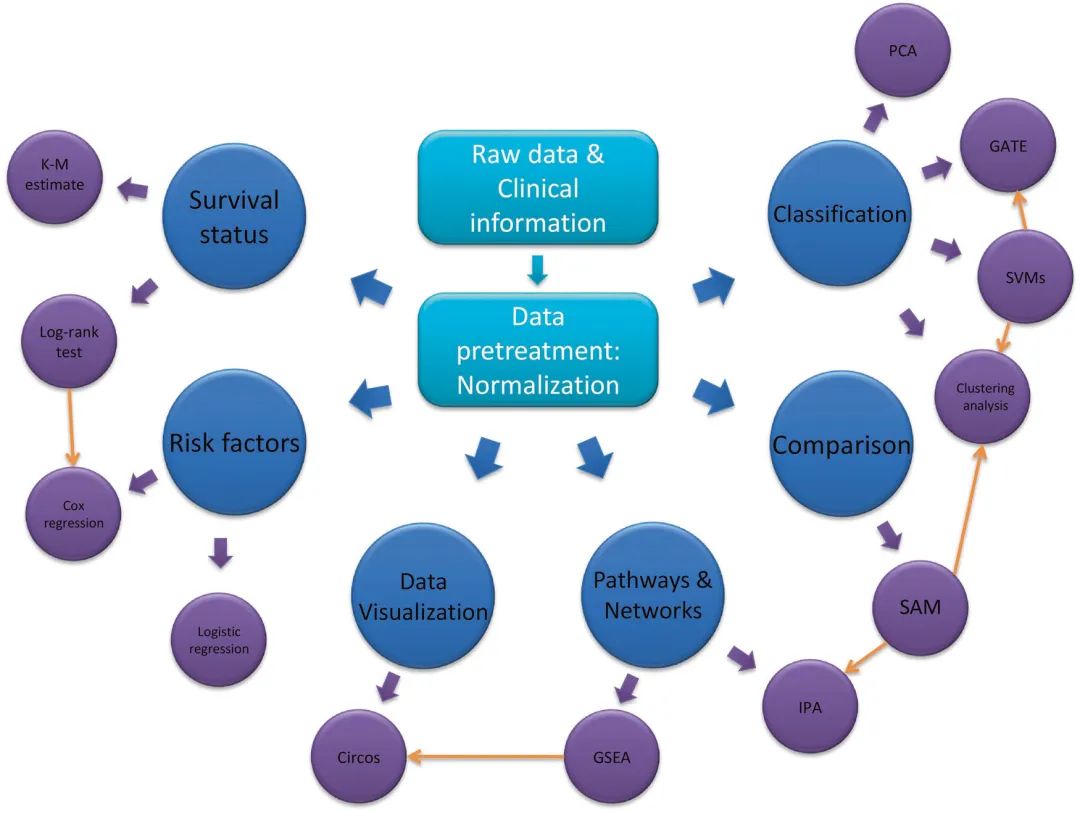
数据分析流程图
归一化
大数据预处理阶段最常见和必要的形式是归一化,用于将所有数据集中并重新缩放为一个整体数值矩阵,以提高其数值稳定性、整体性能和模型拟合。例如,最常用的归一化公式是Z-score,也称为标准分数。
芯片显著性分析(SAM)
SAM是一个Microsoft Excel插件包,是一种广泛使用的基于高通量的互换方法,使用改进的t-statistics (q-value)来识别在蛋白质组数据中多组样本之间的差异表达蛋白,该方法衡量蛋白质丰度和疾病结果之间关系的强度。与小样本量的常规t-test不同,SAM算法非常适用于大数据,通过对蛋白质丰度的列进行置换,并通过最近邻算法自动插补缺失数据,使假阳性和假阴性的数量最小化。此外,SAM的一个有价值的特点是,它利用数据的排列组合给出了错误发现率的估计值,即可能被偶然确定为有意义的蛋白质的比例。
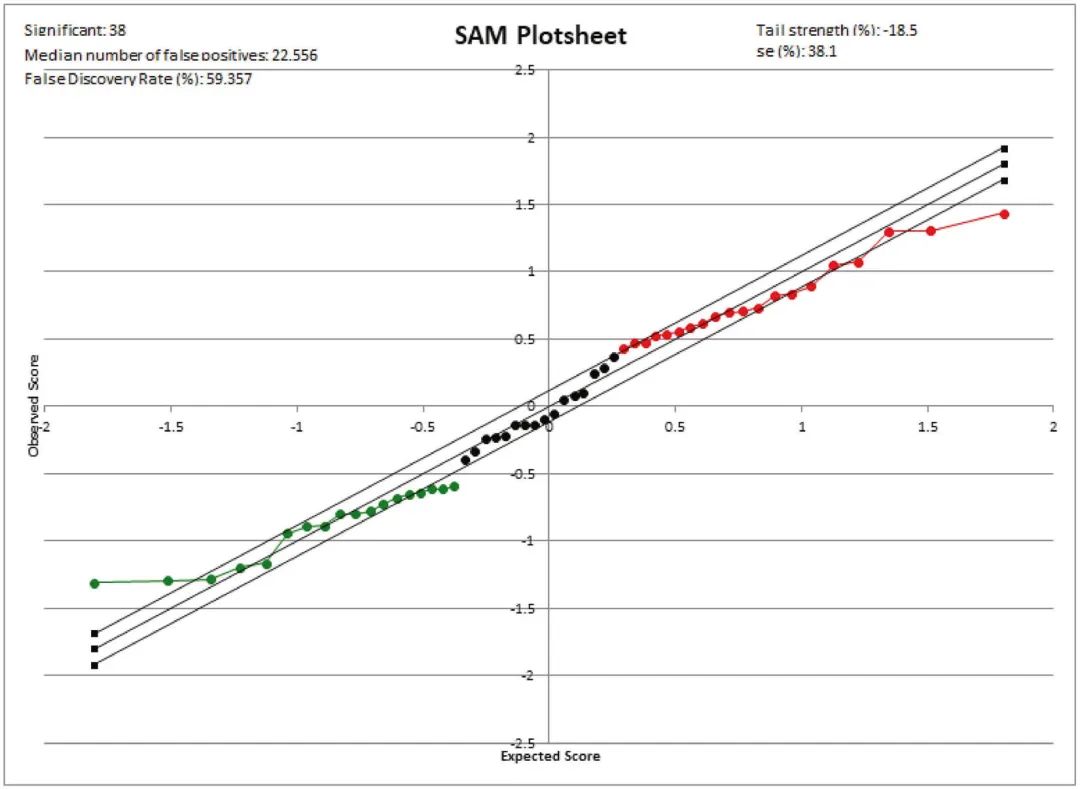
SAM分析生成的图
聚类和判别分析
层次聚类算法(HCA)已被用于通过形成基于数学模型的树状图对大数据进行聚类。为了测量数据点之间的距离,建立了几个基于数学公式的优化模型,包括曼哈顿距离(L1)、欧几里得距离(L2)、皮尔逊相关系数等。距离度量的选择会影响HCA的性能,因此应谨慎决定。在HCA之前,应明确定义基本变量(生物标志物)、样本选择标准和研究目标,以便进行稳健和可重复的分析。此外,HCA可分为单向和双向HCA。还有一种特殊的聚类分析,称为时间序列表达的网格分析(GATE),用于根据时间序列分析和可视化高维生物分子。
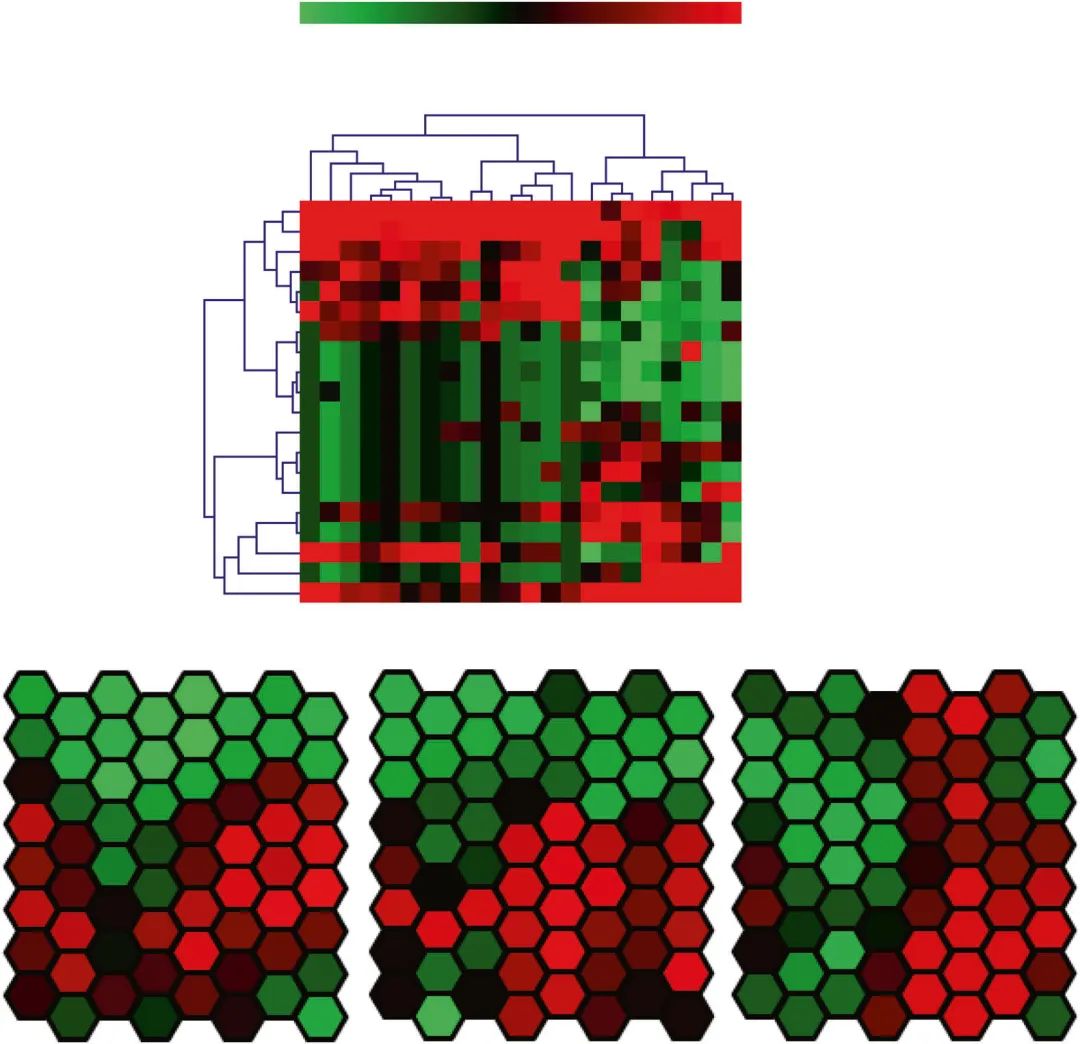
层次聚类分析(HCA)和时间序列表达的网格分析(GATE)示例
与对已知样本进行分类的聚类分析不同,(预测性)判别分析是根据算法在训练集中学习和建立的内容对未知样本进行分类,例如不依赖于数据类型的支持向量机(SVM)可用于线性分离数值或分类数据,并确定潜在的生物标志物作为分类器。(基于机器学习与否)判别分析的主要目的是设计一个计算有效的统计学模型,对多组受试者进行分类,并确定预测率较高的潜在分类器。
生存分析
Kaplan-Meier(K-M)曲线是一种时间事件统计方法,用于研究终点事件与时间周期之间的关系。它可用于评估生存时间、疾病复发、临床试验、动物研究等。K-M估计是计算生存时间的最简单方法。两条生存曲线可以通过log-rank (Mann–Whitney U) 检验进行统计比较,该检验已被广泛使用,包括计算过程中具有不同权重函数的Breslow和Tarone。但它们通常作为单变量分析,不允许测试其他疾病相关变量的影响。相比之下,经常用作多变量分析的Cox比例风险回归模型可以在识别疾病自变量的同时测试其他变量的影响。此外,许多流行的回归模型被用于分析蛋白质组学或基于芯片的大数据,它们的功能在不同程度上类似于Cox回归模型,例如多变量逻辑回归。
主成分分析
主成分分析(PCA)的主要目的是通过创建一组称为主成分的新变量来降低大数据的维数,以表示原始数据集中的大部分信息。因此,只有前几个主成分是最有代表性的,这种每个主成分的变异性逐渐减少的趋势可以用scree图来表示。这种通过主成分降低数据集内维度表示的统计方法对于大数据集或大数据的分类和压缩非常有用。
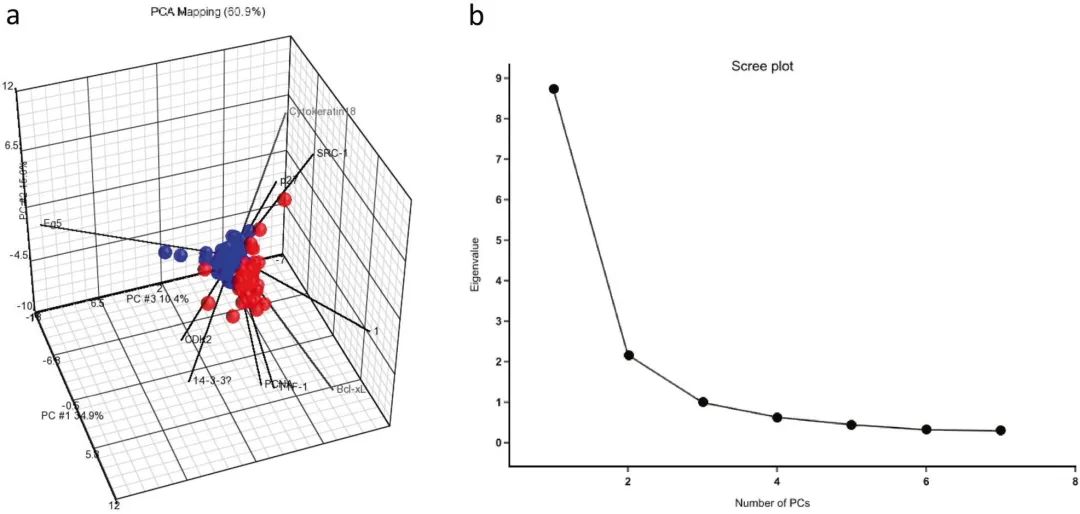
主成分分析
Ingenuity通路分析、基因集富集分析和circos分析
许多分析方法结合在线数据库分析蛋白质组学和微阵列数据,更适合于发现临床意义。Ingenuity通路分析(IPA)是一种基于web的软件应用程序,用于使用表达式数据集进行因果分析。IPA可以同时可视化和分析基因组学、蛋白质组学和代谢组学数据的跨数据库数据,以获得综合各种组学格式的信号网络和典型通路。基因集富集分析(Gene set enrichment analysis, GSEA)是另一种计算方法提供通路富集工具来帮助解释数据集。这种方法关注多个基因作为基因集表达的累积变化,这些基因集共享相似的生物功能、染色体位置或调控,而不是单个基因来识别通路。GSEA方法最显著的优点是它可以捕捉到一些通路,其中几个基因在少量但以一种协调的方式改变。

IPA和GSEA分析示例
此外,Circos是一个软件包,用于在圆形布局中可视化基于组学的数据和信息。可以创建Circos图来探索经典通路与临床病理特征或风险因素之间的关系和贡献。
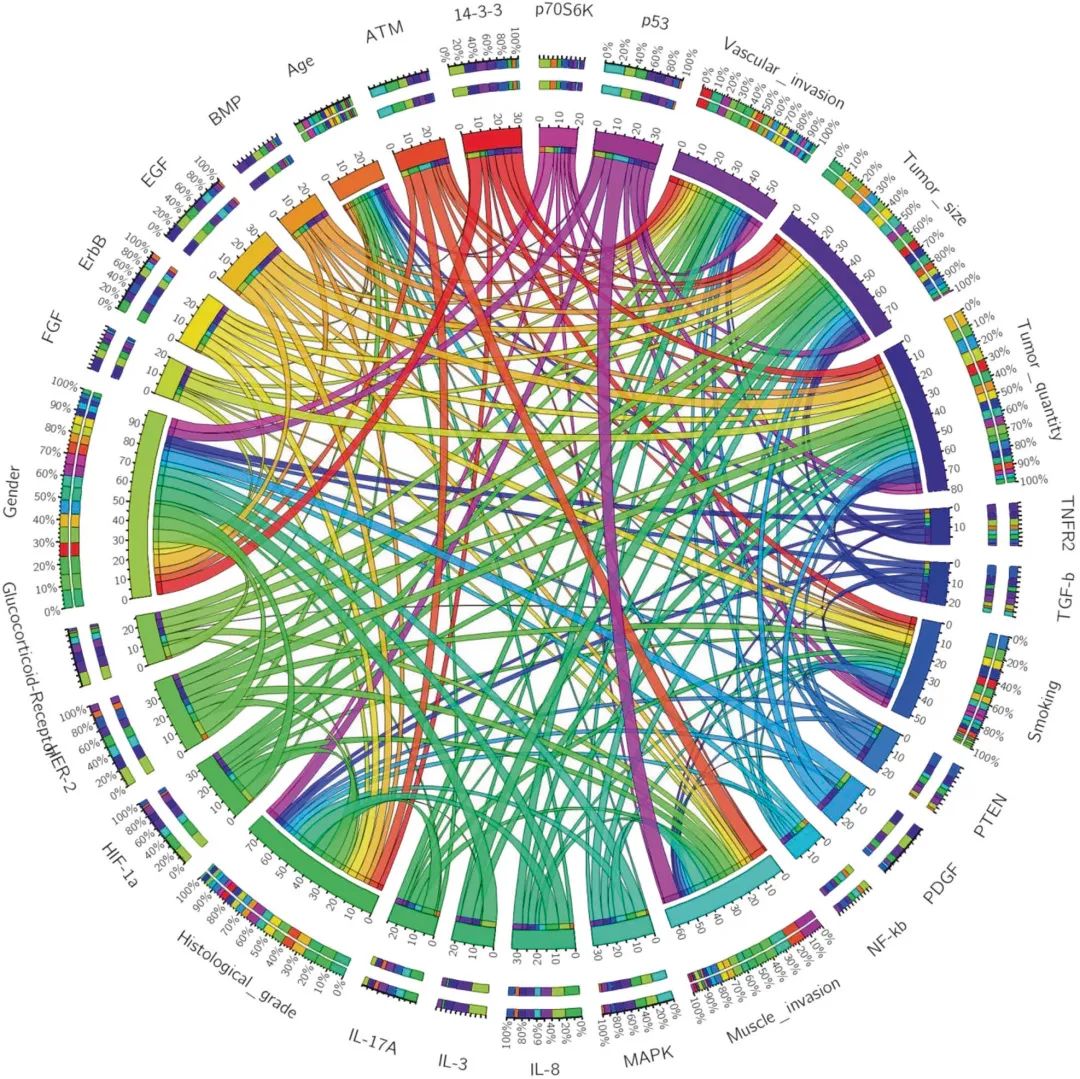
Circos图示例:在所有八种临床病理类型中,性别占分布的最显著比例,表明是对信号网络影响最大的临床因素。
单细胞蛋白组学
SCoPE2和Scp是用于分析多重单细胞蛋白质组数据的R包,而SCeptre是它们在Python中实现的对应软件包。一些通用的蛋白质组学流程也可用于处理单细胞蛋白质组学数据,包括计算质量控制工具和用于数据处理和可视化的单一流程(MSnbase)。
由于篇幅有限,更多技术细节可参考文献原文,点击阅读原文可获取全文 :
https://doi.org/10.1038/s41374-022-00830-7
参考文献
Cui M, Cheng C, Zhang L. High-throughput proteomics: a methodological mini-review[J]. Laboratory Investigation, 2022: 1-12.
图片均来源于参考文献,如有侵权请联系删除。