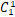点击蓝字 关注我们
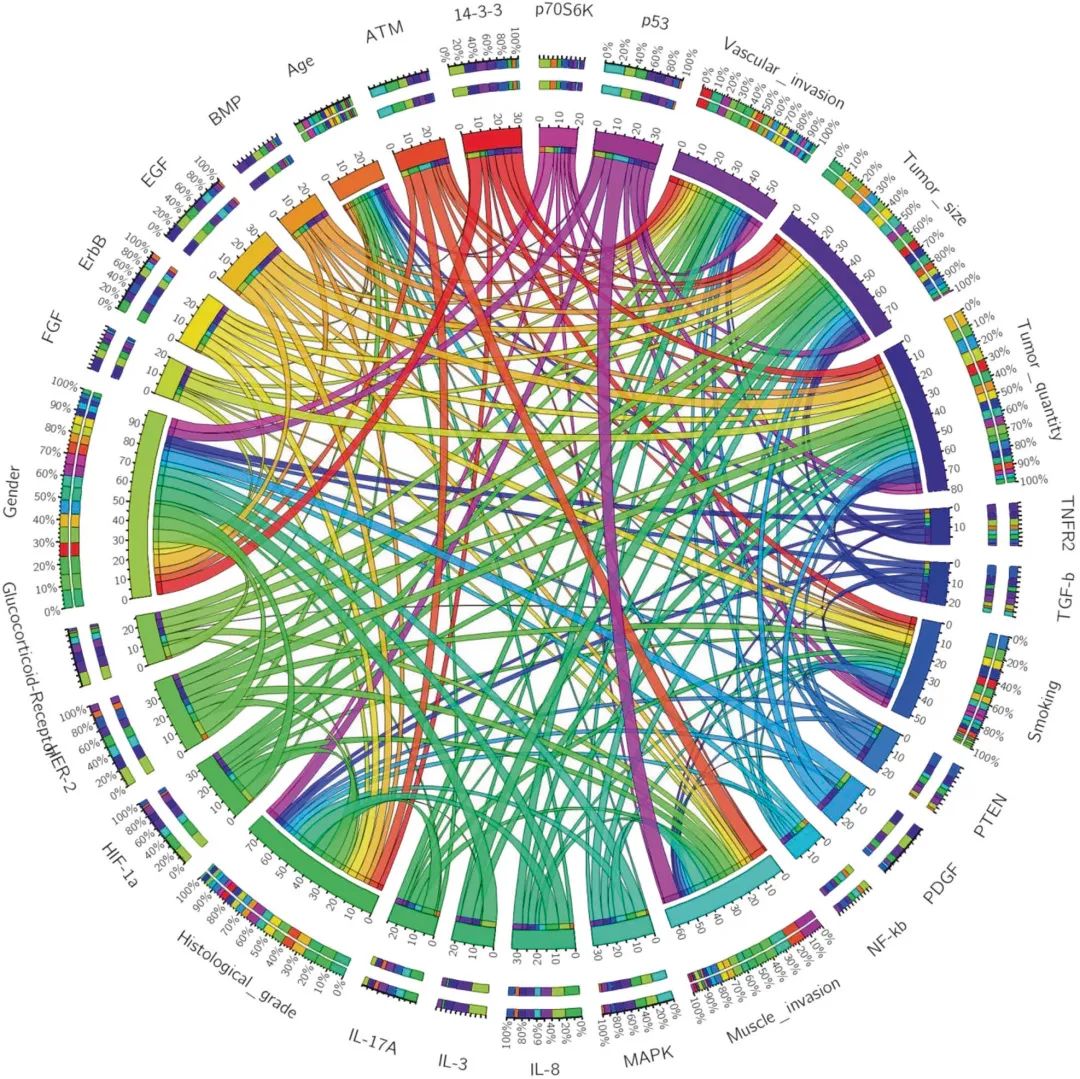
针对微生物组学研究的集成网络分析平台

https://doi.org/10.1002/imt2.13

2022/3/16
● 2022年3月16日,中国科学院生态环境研究中心邓晔组在iMeta在线发表题为“iNAP: An integrated network analysis pipeline for microbiome studies”的方法类文章。
● 该文章发表了一个可用于微生物组学研究、生成和分析生态网络的在线分析平台iNAP,提供多种网络分析方法和工具,可以帮助研究人员更好地了解微生物群落构建机制。
● 第一作者:Kai Feng(冯凯)
● 通讯作者:Ye Deng(邓晔)
(yedeng@rcees.ac.cn)
● 合作作者:Xi Peng(彭玺), Zheng Zhang(张政), Songsong Gu(顾松松), Qing He(何晴), Wenli Shen(沈文丽), Zhujun Wang(王朱珺), Danrui Wang(王丹蕊), Qiulong Hu(胡秋龙), Yan Li(李燕), Shang Wang(王尚)
亮 点
● iNAP针对不同分类学水平的微生物物种,提供了物种域内关联和跨域间物种关联的网络分析流程,即分子生态网络分析平台(MENAP)和跨域生态网络分析平台(IDENAP)
● 为便于微生物组学数据的生态网络分析,iNAP提供了SparCC、eLSA、SPIEC-EASI和基于RMT理论的Pearson/Spearman相关性等构建关联的方法
● iNAP平台免费注册,操作简单,无需生物信息分析和编程知识,即可快速方便的完成网络分析
摘 要
iNAP是可用于微生物组学研究中,生成和分析生态网络的在线分析平台,主要由网络构建和网络分析两个部分组成,同时集成了多个分析工具。网络构建包含多种方法,主要有基于随机矩阵理论(RMT)的相关性方法(Pearson/Spearman),针对组成数据的稀疏相关性(SparCC),局部相似性方法(eLSA )和稀疏逆协方差估计(SPIEC-EASI)。网络分析则提供了网络拓扑结构属性和环境因素与网络结构的关联分析。从微生物组数据到网络分析结果,iNAP包含分子生态网络分析(MENAP)和跨域物种生态网络分析(IDENAP)的完整分析流程,分别对应于多个分类水平下微生物物种的域内和域间互作关系。本文我们以 IDENAP 为例描述了详细流程,并展示利用数据集进行网络分析的全部操作步骤。平台上也提供了从本地分析到在线操作转换的辅助分析工具。因此,iNAP 作为一个提供多种网络分析方法和工具的易于操作的在线平台,可以帮助研究人员更好地了解微生物群落构建机制。
iNAP 可在 http://mem.rcees.ac.cn:8081 免费注册
视频1. iNAP网站进行微生物组网络分析教程
Bilibili:https://www.bilibili.com/video/BV1a3411p72v/
Youtube:https://youtu.be/lCb-Nsp5bwM
视频2. iNAP分析结果采用CytoScape和Gephi可视化教程
Bilibili:https://www.bilibili.com/video/BV1LT4y1i7Sy/
Youtube:https://youtu.be/jAYexCTZYlI
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引言
从宏基因组学数据中解析复杂微生物群落中的物种互作关系,可以更好的理解自然环境和宿主环境微生物的群落构建机制。微生物网络分析可以发现群落中的一些关键物种,在系统抵抗外界扰动或者物种入侵中发挥作用,被广泛用于土壤等复杂生态系统研究中。在群落水平上,网络结构也用于指示环境扰动或者物种丢失时的系统稳定性。不仅仅是物种域内互作关系,不同营养层级的微生物域间物种互作关系,也逐渐引起重视。虽然网络分析在微生物生态学中的应用存在一些问题,但只要使用得当和推测合理,网络分析仍可提供有用信息。
微生物的网络分析多依赖于不同的统计分析方法。基于Pearson或Spearman相关性的方法,MENA网页平台使用RMT理论可以找到一个合适的阈值生成网络。SparCC和SPIEC-EASI的方法也可以用于推断微生物关联,并提供了Python和R的运行脚本。eLSA和LA的方法多用于时间序列的微生物组学数据。基于此,我们开发了网络分析集成平台iNAP,可以用于推断微生物域内及域间物种的关联,并为探究网络拓扑结构属性与环境驱动因子间的关系提供了统计分析工具。本操作手册将展示iNAP的基本流程和个性化分析手段。
平台总览
iNAP平台针对微生物的多度数据,包含分子生态网络(MENs)和跨域生态网络(IDENs)两个分析流程(图1)。对于非时间序列数据集,主要有SparCC、SPIEC-EASI和基于RMT的Pearson/Spearman方法;对于时间序列数据集,主要有eLSA/LA和基于RMT的Pearson/Spearman方法。我们使用邻接网络矩阵(adjacent network matrix)和二分网络矩阵(bipartite network matrix)来指示域内和域间物种的关联。
使用Galaxy的网页框架,iNAP平台为用户提供了便捷的操作流程,无需生物信息学或者编程技能,即可完成微生物网络分析。同时,iNAP也提供了多种计算方法和网络拓扑结构属性的计算(图2)。依据平台上提供的测试文件和本操作说明,每一个用户都可以简单操作并获得相应的结果。由于MENs和IDENs的网络构建流程是相似的,本操作说明主要演示IDENs的操作流程。
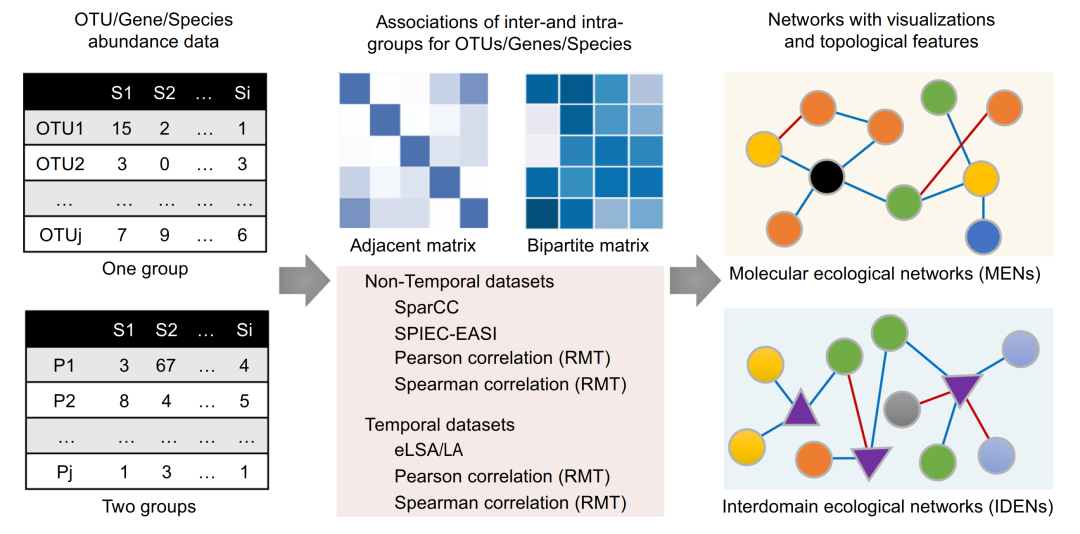
图1:iNAP平台网络分析的简化示意图
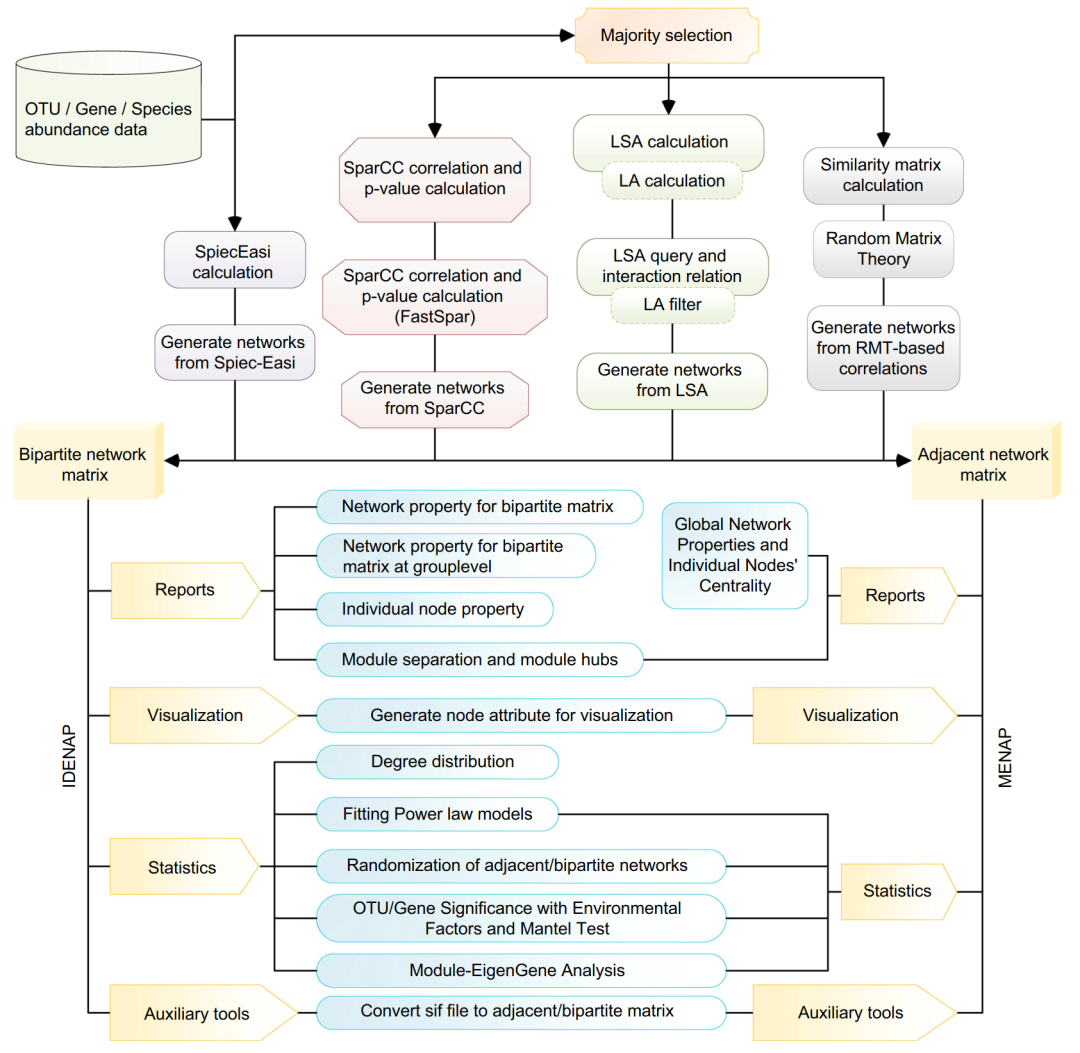
图2:iNAP平台网络构建和分析流程图
网络构建的基本流程
目前,iNAP主要提供四种网络构建方法,在这里,我们主要以跨域物种生态网络分析(IDENAP)为主,对每一个方法单独进行描述。
● 方法1:SparCC
1. Majority selection. 考虑到微生物数据的稀疏性,我们建议对多个样本中的共有物种进行后续分析,比如保留至少一半样本中都检测出的物种。对于IDENAP流程,两个物种类群可以单独设置自己的阈值。经过筛选后,iNAP最低要求有8个样本以及不超过1000个物种的多度数据,否则,系统会提示“Our pipeline cannot process the dataset with larger than 1000 OTUs/genes for this method”。对超过1000个物种的大数据集,我们在Github也提供了本地化运行的建议和流程进行参考(https://github.com/yedeng-lab/iNAP)。
2. SparCC correlation and p-value calculation.输入步骤1的输出文件,使用SparCC的默认参数,可以获得相应的相关性矩阵结果和P值结果。值得注意的是,100可以作为随机次数,适用于大多数的数据,否则计算时间加倍。同时,我们也加入了更快速的计算方法FastSpar,可以处理多于1000个物种的数据。
3. Generate networks from SparCC.基于自己划定的阈值(相关性和P值),只有高于该阈值的物种关系才会被保留,比如SparCC的默认阈值为0.3的相关性和低于0.05的显著性水平。IDNEAP流程中选择bipartite模式,输出文件有Cytoscape和Gephi两种格式(Box 2)。
● 方法2:SPIEC-EASI
4. SpiecEasi calculation.关于majority筛选参数跟步骤1类似,其余参数主要以SPIEC-EASI默认参数为主。输出文件有逆协方差矩阵结果、计算过程报告和矩阵文件。
5. Generate networks from Spiec-Easi.使用步骤4的逆协方差矩阵结果文件,生成网络矩阵和可视化文件(Box 2)。
● 方法3:eLSA
6. Majority selection.同步骤1相同。
7. LSA calculation.针对时间序列数据集,需要指定重复样本数、时间点数和时间延迟点数。其他参数以LSA默认参数为主。输出结果包含物种两两之间的LS值及P值。
8. LSA query and interaction relation.基于LS值、P值或Q值等参数,筛选结果,如“|LS|>=0.28,P<0.001, Q<0.05”,多个筛选标准可以通过“Insert Additional Query Condition”添加。
9. (optional) LA calculation and LA filter.类似于步骤7和步骤8,主要针对LA值计算和 相应的结果筛选。
10. Generate networks from LSA.使用步骤8的文件,输出网络结果(Box 2)。
● 方法4:基于RMT的相关性
11. Majority selection. 同步骤1相同。
12. Similarity matrix calculation.对多度数据中的空值或零值,可以保持不变或填入较小的数值,如0.01,也可以对数值进行对数转换,来计算成对物种之间的Pearson和Spearman相关性。输出文件包含相关系数和P值。
13. Random Matrix Theory.基于随机矩阵理论,从0.01到1以0.01的步长,从相关系数矩阵里头,寻找一个合适的阈值cutoff,表明网络特征基因的分布从泊松分布到高斯正交分布转变。输出结果有阈值和对应的χ2检验结果,挑选P值在0.05以上的cutoff值,作为网络构建的阈值。
14. Generate networks from RMT-based correlations.依据步骤13的阈值,生成网络(Box2)。
网络分析流程
15. Network property for bipartite matrix. 输入文件为矩阵文件,文件输出包含网络整体的拓扑结构属性,详细介绍在iNAP页面。
16. Network property for bipartite matrix at group level. 输入文件为矩阵文件,文件输出包含网络两个类群水平上的拓扑结构属性,详细介绍在iNAP页面。
17. Individual node property. 输入文件为矩阵文件,文件输出包含网络物种水平的拓扑结构属性,详细介绍在iNAP页面。
18. Module separation and module hubs. 输入文件为矩阵文件,文件输出包含模块划分结果和基于模块所计算的节点模块内连接度与模块间连接度(Z-P值),基于Z-P值,可以将物种在模块间的作用划分为:网络核心、模块内核心、模块间核心和普通节点。
19. Generate node attribute for visualization.输入文件为步骤17和18的结果,输出对应Cytoscape或Gephi格式的节点属性文件。
20. Degree distribution. 输入文件为矩阵文件,在两个物种类群水平上,对节点的连接度分布进行拟合,主要有指数分布、幂律分布和截断幂律分布。
21. Fitting Power law models.同步骤20类似,将所有物种综合考虑,拟合节点的连接度分布,主要有指数分布、幂律分布、指数幂律分布和截断幂律分布。
22. Randomization of bipartite networks. 输入文件为矩阵文件,生成100个随机网络并计算拓扑结构属性,输出结果包含均值和标准偏差。
23. OTU/Gene Significance with Environmental Factors and Mantel Test.使用步骤1的文件、网络矩阵文件和新上传的环境属性文件,计算网络节点与环境属性的相关性;基于相关系数和网络节点的连接度属性,进行mantel检验。
24. Module-EigenGene Analysis. 使用步骤1的文件、新上传的环境属性文件和步骤18的文件,计算模块特征基因和环境属性间的关系。
辅助工具
25. Convert sif file to bipartite matrix. 该步骤主要是将sif文件转换为二分网络矩阵。主要是根据ID标签进行区分两个物种类群,如“B_OTU”和“P_OTU”。
26. Taxonomy summary of low-level species for bipartite networks. 根据微生物的物种分类注释信息,将二分网络矩阵在门或纲水平上拆分成多个子网络。
邻接矩阵网络的简要流程
对于MENAP,由于邻接网络矩阵只有一个物种类群,步骤1中需要选择“one group of species”,在网络构建完成后,使用以下流程完成网络分析。
27. Global Network Properties and Individual Nodes' Centrality. 输入文件为矩阵文件,文件输出包含网络整体和节点的拓扑结构属性,详细介绍在iNAP页面。
28. Module separation and module hubs. 输入文件为矩阵文件,文件输出包含模块划分结果和基于模块所计算的节点Z-P值,同步骤18相同。
29. Generate node attribute for visualization 输入文件为步骤17和18的结果,输出对应Cytoscape或Gephi格式的节点属性文件。
30. Fitting Power law models. 输入文件为矩阵文件,拟合节点的连接度分布,包括指数分布、幂律分布、指数幂律分布和截断幂律分布。
31. Randomization of adjacent networks 输入文件为矩阵文件,生成100个随机网络(节点连接度不变)并计算拓扑结构属性,输出结果包含均值和标准偏差。
32. OTU/Gene Significance with Environmental Factors and Mantel Test 使用步骤1的文件、步骤28的模块属性文件和新上传的环境属性文件,计算网络节点与环境属性间的相关性系数;基于相关系数和网络节点的连接度属性,进行mantel检验。
33. Module-EigenGene Analysis. 使用步骤1的文件、步骤28的模块属性文件和新上传的环境属性文件,计算模块特征基因和环境属性间的关系。
34. Convert sif file to adjacent matrix. 转换Cytoscape的sif格式文件为邻接网络矩阵,进行步骤27到33的分析。
Box1 输入文件
通常情况下,iNAP的输入文件主要以txt或者tabular格式的文本文件为主。文件的列名和行名通常对应样本名和物种编号,建议使用“字母+数字”的组合并以字母开头的方式进行命名,如S1、P1和OTU1,而不是1S、1P和1OTU。其他特殊字符(如空格、星号、警号等)都以下划线代替。由于iNAP使用“_M”和“_P”来区分两个物种类群,在输入文件命名时,最好避开类似的命名方式。示例文件已存储于iNAP网站的共享数据库。
多度数据集(Abundance datasets, .txt/.tabular):该数据集为不同样本的物种多度数据,数据行为物种信息,数据列为样本信息,列名以“OTUID”、“OTU”或者“制表符”开始。
属性文件(Metadata files, .txt/.tabular):该文件主要包括样本的环境属性,数据行名是样本信息,数据列名是环境属性信息。
分类信息文件(Taxonomy files, .txt/.tabular):该文件主要用于展示物种编号的分类学信息,通常包括域、门、纲、目、科、属和种等信息。
Box2 输出文件
网络构建会生成邻接或二分网络矩阵和对应的可视化文件(图2)。对于IDENAP,通常包括四个网络类别:整体网络、域间网络和两个类群的内部连接网络。对于MENAP,则只有一个邻接网络的结果。可视化的输出文件,提供Cytoscape和Gephi软件所对应的两种文件类型(图3)。
邻接网络矩阵:该矩阵是一个方阵,行名和列名相同且对角线上的数值为0。矩阵中的1或0表示连接的有无。
二分网络矩阵:该矩阵通常包含两个物种类群,行名和列名之间没有重复。矩阵中的1或0表示连接的有无。
Cytoscape文件:包含三个文件,即网络文件(.sif),节点属性文件(node attribute file, .txt)和连接属性文件(edge attribute file, .txt)。
Gephi文件:包含两个文件,即节点属性文件(node attribute file, .csv)和连接属性文件(edge attribute file, .csv)。
图3:使用Cytoscape(A)和Gephi(B)绘制网络。节点颜色表示不同的物种类别(A)和模块区分(B)
iNAP的特点和应用
iNAP平台依据“SAFE”模式进行设计与维护,具有稳定、开放、灵活和简单等特点。我们将支持平台的运行与定期维护,保证正常访问。利用平台上提供的多种分析工具,用户可以灵活选择上传原始数据或本地运行的临时输出文件,进行后续分析。iNAP也支持多个方法之间的交叉使用,例如SparCC的相关性结果可以基于RMT理论来寻找合适的阈值。将这些分析工具整合进网页框架,用户可以参照本操作说明,便捷的分析数据。iNAP网络平台,使得探究微生物群落的构建机制和寻找介导生态系统稳定性的关键物种,更加简便。
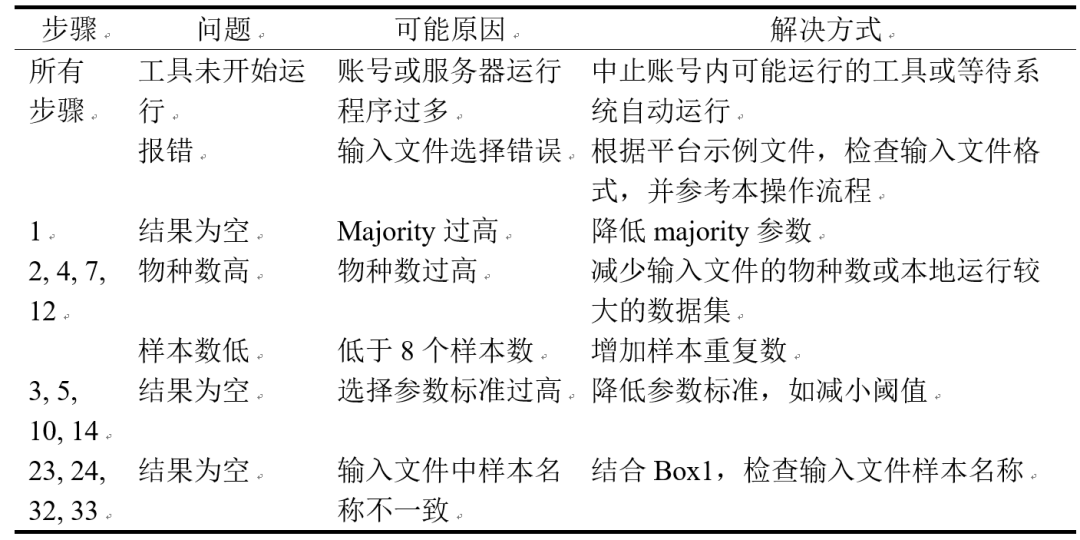
表1 排除运行错误的建议
数据开放性
iNAP平台(http://mem.rcees.ac.cn:8081)对所有研究人员开放使用,用户可以免费快速注册。平台上所有的分析工具都可匿名访问,为便于平台管理和解决问题,我们鼓励用户使用注册账号进行分析。同时,我们在Github(https://github.com/yedeng-lab/iNAP)也提供了本地化运行SparCC、LSA和SPIEC-EASI的计算流程。
引文格式:Feng, Kai, Xi Peng, Zheng Zhang, Songsong Gu, Qing He, Wenli Shen, et al. 2022. “iNAP: an integrated Network Analysis Pipeline for microbiome studies.” iMeta 1: e13. https://doi.org/10.1002/imt2.13
作者简介

冯凯
● 中国科学院生态环境研究中心特别研究助理/博士后研究人员
● 2020年毕业于中国科学院大学,获理学博士学位。从事微生物生态学和生物信息学研究,目前主要关注气候变化下的微生物地理分布格局和微生物群落构建机制。以第一和共同第一作者在Science Bulletin,Molecular Ecology Resources,Molecular Ecology,Frontiers in Microbiology等期刊发表论文8篇,合作发表论文30余篇。

邓晔(通讯作者)
● 中国科学院生态环境研究中心研究员,中国科学院大学资源与环境学院博士生导师。中国科学院“百人计划”终期评估优秀,国家计划青年拔尖人才
● 研究工作围绕环境微生物群落生态学开展,通过一系列的分子检测工具和数据分析手段的开发,对全球气候变化下微生物的响应和反馈机制以及微生物的地理学分布格局开展了一系列创新性的工作。迄今在各类SCI杂志上(包括Science、Ecology Letters、Nature Climate Change、Nature Communications, PNAS等)已发表研究论文200余篇,总引用15000余次,H-index为61。2018-2021连续四年获得科睿唯安“全球高被引学者”。现任中国生态学会学会微生物生态专业委员会秘书长、国际微生物生态学会中国青年大使。担任ISME Journal资深主编、Microbiome执行编辑、Journal of Plant Ecology、Soil Ecology Letters等期刊编委。
更多推荐
(▼ 点击跳转)
iMeta第一期免费订阅开始啦
iMeta期刊纸质版开始免费订阅(包邮)——第一期创刊收藏版

iMeta文章中文翻译+视频解读
iMeta | 南科大宋毅组综述逆境胁迫下植物向微生物组求救的遗传基础(附招聘)
▸▸▸▸
iMeta:青岛大学苏晓泉组开发跨平台可交互的微生物组分析套件PMS

▸▸▸▸
iMeta:德布鲁因图在微生物组研究中的应用
▸▸▸▸
iMeta:哈佛刘洋彧等基于物种组合预测菌群结构的深度学习方法
▸▸▸▸
iMeta:吴青龙/王明福/刘金鑫等-从肠道菌群看待人类对高原饮食的适应性
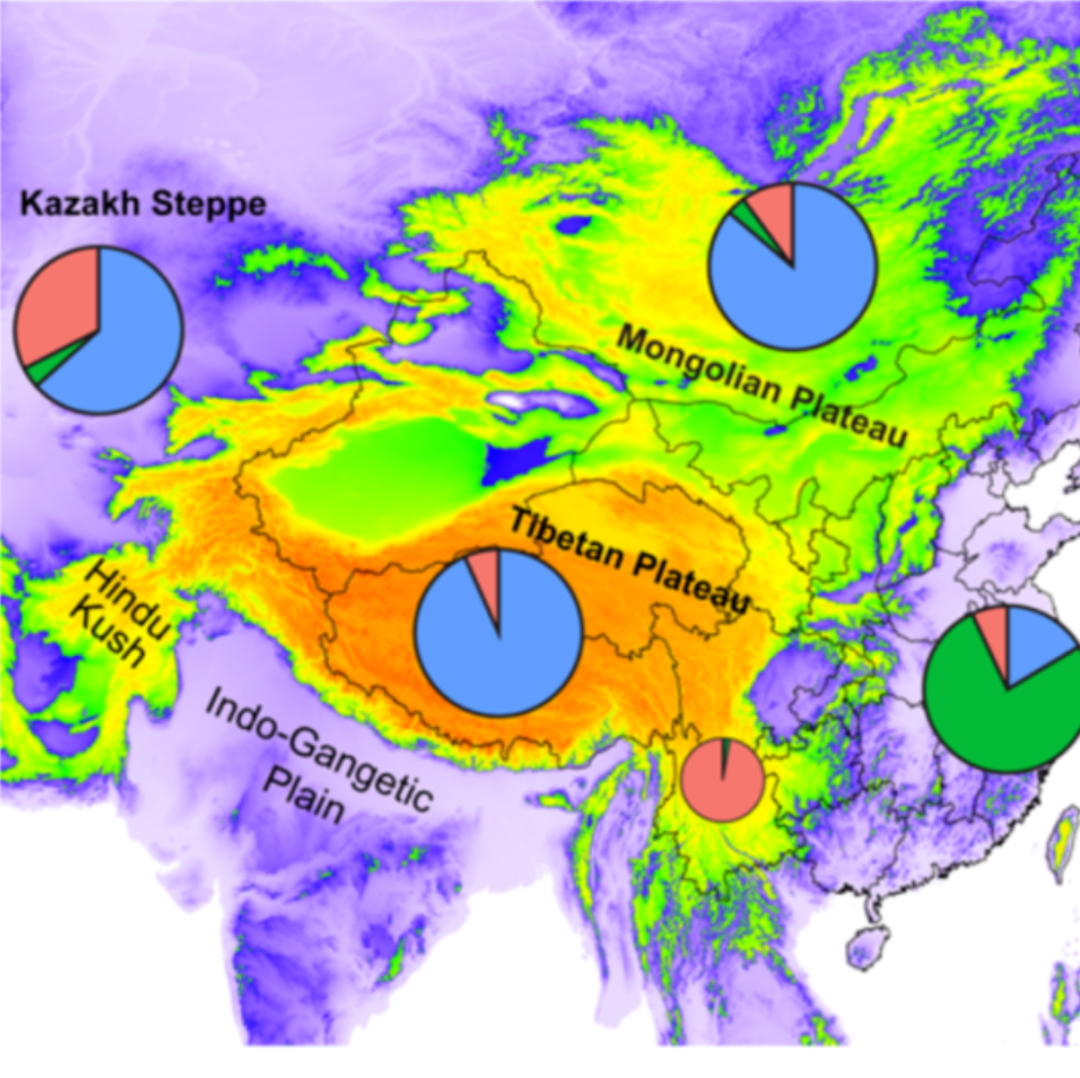
▸▸▸▸
iMeta:西农韦革宏团队焦硕等-土壤真菌驱动细菌群落的构建
▸▸▸▸
iMeta:高颜值高被引绘图网站imageGP
iMeta教你绘图
使用ImageGP绘图热图Heatmap
▸▸▸▸
使用ImageGP绘图富集分析泡泡图
期刊简介

“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science

微信公众号
iMeta
责任编辑
微微