读读论文,用谷歌学术翻译一下,重要的部分做一下笔记。正文部分是翻译,加黑部分是个人笔记。
本次学习的论文:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5468393/
1. 摘要
“缺失遗传力”问题表明,全基因组关联研究中的遗传变异不能完全解释复杂性状的遗传力。传统上,表型的遗传力是通过对双胞胎、兄弟姐妹和其他近亲的家族研究来衡量的,并假设他们之间的遗传相似性。当将该遗传力与通过GWAS获得的相同性状的遗传力进行比较时,两个测量值之间出现了很大差距,全基因组研究报告的值明显较小。已经提出了这种“缺失遗传力”的几种机制,如表观遗传学、上位性和测序深度。然而,它们都不能完全解释这种遗传力差距。在本文中,我们提供的证据表明,为了广泛理解和解释人类特征的表型遗传力,必须考虑人类微生物组的组成和功能多样性。这一假设基于以下几个观察结果:(A)人类微生物组的组成与许多重要特征有关,包括肥胖、癌症和神经系统疾病。(B) 我们的微生物组编码的第二个基因组的基因数几乎是人类基因组的100倍,这第二个基因可能是遗传变异和表型可塑性的丰富来源。(C) 人类基因型与我们微生物组的组成和结构相互作用,但其本身无法解释微生物变异。(D) 微生物的遗传组成会受到宿主行为、环境或其他宿主的垂直和水平传播的强烈影响。因此,家族研究中假设的遗传相似性可能会导致遗传力值的高估。我们还提出了一种方法,允许将我们微生物组的组成和功能多样性纳入全基因组关联研究。
遗传的遗传力,真实的遗传力,是通过双胞胎、兄弟姐妹相关的家系信息就行评估的。GWAS分析得到的显著SNP得到的遗传力,这一部分的差异,就是消失的遗传力。之前的挖掘点,在于群体大小、挖掘多个变异来源。本篇介绍微生物的影响,包括:微生物的影响、微生物和人类互作。
2. GWAS介绍和缺失的遗传力
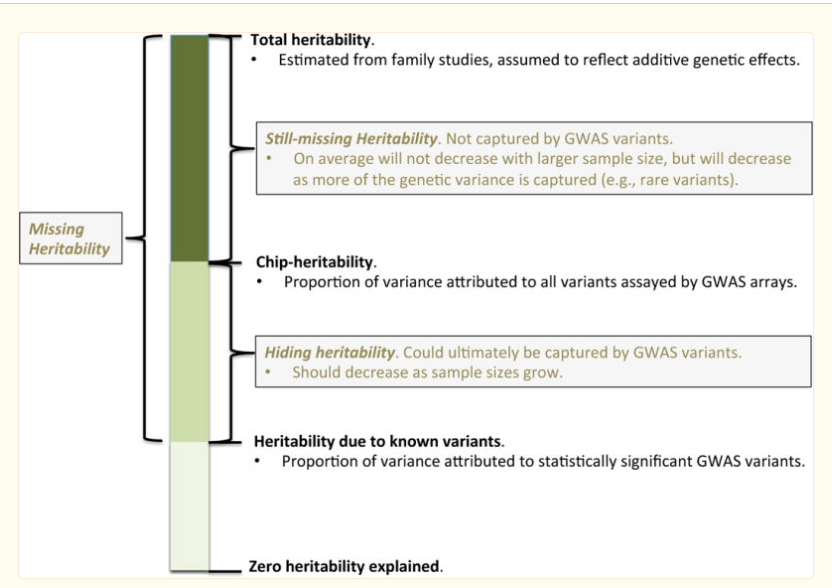
表型的广义遗传力(H2)定义为可由遗传方差解释的表型变异比例。十年前,遗传变异几乎不可能精确测量,通常是从亲属关系假设的。假设父母和子女之间有50%的基因同一性,就像第一个兄弟姐妹一样,而同卵双胞胎则被认为具有完全同一性。这些研究主要基于系谱数据,因此遗传力估计总是包括所有因果变异的贡献,为了计算性状的遗传力,需要做出一些假设(Visscher等人,2008)。如今,随着全基因组关联研究(GWAS)的出现,可以根据从无关个体群体中收集的单核苷酸多态性(SNP)来估计性状的遗传力。为了估计一个性状的狭义遗传力,这些研究收集了数千个遗传变异的信息,并通过遗传身份计算任何两个个体之间的关联度。狭义遗传力(h2)定义为可由遗传线性效应解释的表型变异比例,由于GWAS与单个SNP相关,因此它提供了这种类型遗传力的估计。到今天为止,我们知道超过50000个SNP与许多重要的人类表型相关。然而,这些SNP的个体效应和累积效应都无法解释其相关表型的遗传力(Lee等人,2011)。例如,系谱研究表明,人类身高的80%变异来自遗传效应。GWAS研究发现了大约50种与人类身高相关的遗传变异,但它们只能解释5%的身高变异。这两种测量值之间的差异出现在许多人类特征中,被称为缺失遗传力问题。旨在寻找其来源的努力仍在进行中(Manolio等人,2009年;Eskin,2015年)。
对于这种缺失的遗传力隐藏在哪里,有许多可能的解释,但没有达成共识。表观遗传学、基因相互作用、RNA、遗传力高估、小尺寸效应变体、GWAS实验局限性和许多其他因素被认为是这个问题背后的可能原因(Slatkin,2009;Marian,2012;Zuk等人,2012;Grandjean等人,2013)。然而,我们仍然无法解释人类特征的完全遗传性。在GWAS中,当几个SNP与给定表型在显著水平上相关时,就会出现一个经常报告的问题,因为这些变体通常具有较小的效应大小。这意味着,尽管许多变体可能与单个特征显著相关,但拥有其中任何一个变体都不会显著增加发展该特征的几率。一个例子是人类中的LMTK2变体。尽管与前列腺癌有显著相关性,但这种特殊变体的存在并不会增加患者罹患前列腺癌的几率(Zuk等人,2012年)。为了避免其中一些问题,已经提出了候选基因研究的更新版本,其中他们选择对先前通过GWA识别的特定基因进行深度序列分析,而不是筛选整个基因组。这种方法具有更高的分辨率,并有助于检测新的强效应变体(Zuk等人,2014;Tsai等人,2015)。然而,这些方法仍然不能为缺失遗传性问题提供明确的解决方案。
GWAS挖掘的位点,效应整体来说也很小,有种思路是将基因相关的序列都进行分析,而不是分析单个SNP,这样效应应该大一些。
为了使本文内容完整,在以下几节中,我们首先简要讨论了当前基因关联研究的一些主要局限性,然后回顾了当前对微生物组的理解及其对我们生理学的重要性。最后,基于这些讨论,我们提出了一个主要假设,即通过考虑微生物组的遗传和功能多样性,可以显著缩小GWAS测量的遗传力与家族研究之间的现有差距,这仍然是被忽视的表型变异来源(Blanco-Gómez等人,2016)。我们还提供了如何执行此计算的一般观点。
3. GWAS的限制和基因互作
GWAS经常做出的一个简化假设是,环境因素(如行为、饮食和疾病)在其受试者中是同质的。这一假设通常不符合。研究表明,不同人群的饮食和运动差异很大,并对肥胖和糖尿病等疾病的发展产生重大影响(Pan等人,1997年)。习惯可能在计算这些表型的遗传力中发挥重要作用,因为行为相似的人可能在基因上不同,但表现出密切的表型相似性。
GWAS也忽视上位性(基因-基因相互作用)和表观遗传效应。基因调控网络的研究表明,基因、蛋白质、RNA和其他调控分子之间的相互作用对特定基因表达模式的产生和维持至关重要。这些模式反过来决定了表型。GWA通常报告与特定特征相关的单个SNP,但SNP可能具有不一定是线性的组合效应。一组遗传变异结合在一起可以产生协同或拮抗作用(Wei等人,2014)。例如,两个SNP的同时出现可能会产生非常强烈的积极影响,当只有一个SNP存在时,这是无法观察到的,但如果还存在第三个拮抗变体,则积极影响可能会受到阻碍,最终结果可能是三个遗传变体之间存在轻度关联。由于SNP集之间也可能发生相互作用(McKinney和Pajewski,2012),因此开发能够检测并考虑这些方案的统计、数学和计算工具对于确定上位效应的重要性至关重要。然而,我们应该注意到,从简单的概率角度来看,上位性往往与生理有关,而不是与遗传力有关,因为其父母的基因混合导致后代的基因相关性丧失(Young和Durbin,2014)。
表型变异的另一个来源是表观遗传修饰。尽管由于几乎所有的表观遗传标记都是通过重编程在胚胎中去除的,因此它们大多被丢弃,但表观遗传标志是否可以跨人类世代传播的问题仍在争论中(Slatkin,2009)。已知甲基化、乙酰化和mRNAs会影响基因表达(Delcuve等人,2009),这一事实对某些疾病的发展具有重要影响。但是,除非这些标记经过几代保存,否则它们在缺失遗传力问题中的意义就不会显著。由于GWA忽略了这些表观遗传标记,我们仍然不知道表观基因组是否被传递以及在何种程度上被传递,因此很难评估它们在人类性状遗传力中的作用。然而,在估计性状的遗传力时,有一个很大的潜在表型变异来源没有被考虑在内。
作为生态适应系统的人类越来越明显的是,如果我们要了解生物学的许多方面,就需要考虑生活在我们体内和我们身上的微生物群落的影响。我们体内约有3.9×1013个微生物细胞(Sender等人,2016年),几乎每个部分都有不同的微生物组成和丰度(Morgan等人,2013年;Blekhman等人,2015年)。这些微生物以多种方式与我们的代谢相互作用,了解这些微生物的精确功能和含义是一项复杂的工作。
微生物组对人类生物学的影响鼓励了文献中引人注目的说法,例如称我们的微生物组为“额外器官”,或将人体称为“超级有机体”(Baquero和Nombela,2012)。然而,这些命名可能会产生误导,因为器官是由具有相同基因组的细胞组成的,超有机体表示同一物种个体的优生群体(Bordenstein等人,2015)。holobiont一词更准确、更有用,因为它表示由许多不同物种的基因组组成的更具活力的实体。当它适应新的环境时,它的组成可以在时间和空间上发生变化。此外,真核细胞的核基因组通常通过经典的孟德尔框架垂直传播,但全息生物的微生物可以垂直和水平传播。例如,可以通过直接亲本转移或稳定的环境传播获得特定的共生微生物,如乳酸杆菌(Powell等人,2014)。与其他人的简单互动,如亲吻或触摸,也会导致微生物转移,从而改变全息生物的组成(Kort等人,2014)。在下一节中,我们认为忽视微生物组的丰富动态及其相互作用可能会阻碍我们对人类表型多样性的理解。
4. 作为生态适应系统的人类
越来越明显的是,如果我们要了解生物学的许多方面,就需要考虑生活在我们体内和我们身上的微生物群落的影响。我们体内约有3.9×1013个微生物细胞(Sender等人,2016年),几乎每个部分都有不同的微生物组成和丰度(Morgan等人,2013年;Blekhman等人,2015年)。这些微生物以多种方式与我们的代谢相互作用,了解这些微生物的精确功能和含义是一项复杂的工作。
微生物组对人类生物学的影响鼓励了文献中引人注目的说法,例如称我们的微生物组为“额外器官”,或将人体称为“超级有机体”(Baquero和Nombela,2012)。然而,这些命名可能会产生误导,因为器官是由具有相同基因组的细胞组成的,超有机体表示同一物种个体的优生群体(Bordenstein等人,2015)。holobiont一词更准确、更有用,因为它表示由许多不同物种的基因组组成的更具活力的实体。当它适应新的环境时,它的组成可以在时间和空间上发生变化。此外,真核细胞的核基因组通常通过经典的孟德尔框架垂直传播,但全息生物的微生物可以垂直和水平传播。例如,可以通过直接亲本转移或稳定的环境传播获得特定的共生微生物,如乳酸杆菌(Powell等人,2014)。与其他人的简单互动,如亲吻或触摸,也会导致微生物转移,从而改变全息生物的组成(Kort等人,2014)。在下一节中,我们认为忽视微生物组的丰富动态及其相互作用可能会阻碍我们对人类表型多样性的理解。
5. 微生物组对人类特征的影响
微生物群对人类健康有很大影响(Cho和Blaser,2012;Clemente等人,2012;Dave等人,2012年;Huttenhower等人,2012)。众所周知,人体的细菌细胞数量与人类细胞数量大致相同(Sender等人,2016年),就基因含量而言,微生物群的基因数量至少是我们自身细胞的100倍(Qin等人,2010年)。微生物遗传成分的最大多样性存在于我们的肠道中,其中细菌细胞占优势(每克结肠组织约1012个细菌细胞,Collins,2014)和大约800-1000种不同的细菌(Bäckhed等人,2005)。这些发现改变了“一种疾病——一种微生物”的传统观点,因为许多与微生物组相关的疾病现在被认为是微生物群落失衡的结果,更广为人知的是失调。这意味着疾病不仅源于某些物种的存在,还源于它们的缺失、相对丰度和/或相互作用(Petersen和Round,2014)。例如,众所周知,微生物群落的破坏在肠易激综合征(IBD)、肥胖和糖尿病等疾病的进展中起着重要作用。相反,肠道微生物组的高度多样性和时间稳定性是健康的重要特征。克罗恩病和衰老等有害状态通常与低多样性有关(Dicksved等人,2008年)。当然,对于幽门螺杆菌等疾病的进展和胃癌的发展至关重要的特定细菌菌株的例子也有,但它们比以前认为的要少见(Perry等人,2006年;Spor等人,2011年)。
微生物组学研究目前面临的主要挑战之一是确定某些微生物物种的存在或缺失对疾病发展的重要性。微生物群中物种的丰度并不总是其在某一特性的发展或存在中的重要性的指标。微生物群中看似无害的初始不平衡可以通过特定病原体的作用进一步放大,这些病原体的丰度可能非常低。例如,在牙周炎症中,已经证明某些关键病原体可以通过禁用免疫反应来放大整个微生物群落的毒力(Lamont等人,2015)。除了这些天生的病原体之外,我们知道某些细菌物种可以突然成为病原体。这些被称为致病微生物的微生物在正常微生物群中相对常见,但在某些条件下,例如宿主体内轻微失去稳态,它们会成为不平衡的放大因子。通过促进宿主炎症的过程,或通过产生细菌素,这些病原菌可以促进微生物群落中其他菌株的致病性,这自然会导致进一步和更强的破坏(Cho和Blaser,2012)。
6. 塑造我们的微生物群
微生物群和宿主之间的相互作用是双向的,因为宿主也影响其微生物群的发育和稳定性。首先,宿主饮食和营养状况的变化可以改变其微生物组成和行为。众所周知,完全由动物产品组成的饮食可以增加耐胆汁微生物的数量,如嗜胆汁菌和拟杆菌,而植物性饮食可以增加代谢植物多糖的厚壁菌的数量。这些变化可以在一周内观察到。除了改变饮食外,在食物或临床治疗中接触抗生素也会对微生物群的功能和组成产生快速而深刻的影响。众所周知,成年人的微生物组组成相对稳定,但抗生素,尤其是广谱抗生素,可以杀死整个共生微生物群落,也可以使抗生素耐药基因的丰度达到峰值(Yassour等人,2016)。为期5天的环丙沙星疗程会降低整体细菌多样性,并改变肠道微生物群中30%物种的丰度(Dethlefsen等人,2008年)。抗生素同样影响非致病性(共生)微生物,通常与我们代谢过程的正确功能和免疫系统的发育有关。它们的消除可能导致代谢改变和免疫反应故障,进而可能导致微生物群中的额外失衡。这可能导致肠道炎症等致病环境,增加肠道感染的易感性,以及进一步失衡。由于物种之间的相互依赖性,某些菌株(或特定功能)的去除也可能产生级联效应,其中未受抗生素影响的微生物本身同样会灭绝,因为它们依赖于其他物种的存在。这种相互联系是微生物组非线性动力学背后的一个原因(Foster等人,2008;Cho和Blaser,2012)。
除了临床应用外,抗生素在养牛方面也有多种应用。通常给家畜施用亚致死剂量的抗生素,不仅是为了防止感染,也是为了促进生长。目前已知,观察到的动物体重增加与微生物群代谢能力和结构的变化有关(Cho等人,2012年)。此外,在这些环境中强烈选择了抗生素耐药菌株,使得艰难梭菌等多重耐药病原体非常普遍。事实上,即使在去除药物后,这种病原体的感染仍会在人类宿主中持续存在(Chang等人,2008年;Manges等人,2010年)。最近的证据表明,在去除抗生素后,抗生素使用的反弹可能持续数周甚至数月(Dethlefsen等人,2008年)。根据元基因组研究,在接触头孢丙烯一周后,人类粪便样本中出现了之前未检测到的耐药基因(Raymond等人,2016)。遗传水平转移在微生物多样性中也发挥着重要作用,因为它可以触发许多以前易感菌株中抗性基因的快速获取。因此,在临床和农业环境中过度使用抗菌剂与全球耐药菌株的发病率增加有关并不奇怪(Raymond等人,2016年)。
微生物,抗生素,杀菌和促进生长。
7. 水平和垂直微生物传播
2013年,艰难梭菌感染(CDI)被美国疾病控制和预防中心列为抗生素耐药性报告中的紧急威胁。CDI的复发率约为15%-30%,这意味着患者需要解除状态、营养不良和再次住院,以及其他不便。目前的治疗主要基于抗生素,成功率在30%到80%之间,复发性感染更难治疗。然而,整个微生物群落的移植已显示出控制这种病原体生长的巨大潜力(Khoruts等人,2009年)。这些微生物移植包括将整个微生物群落从一个个体转移到另一个个体,通常是从成人粪便样本。通过重新引入可能缺失的物种,该技术旨在恢复肠道微生物群的平衡,并减少任何可能由病原体首先促进的失调。如最近所示,经机器翻译后,感染患者的微生物组组成与健康个体的微生物组构成非常相似(Weingarden等人,2014),表明机器翻译可以稳定地改变患者的微生物组成。使用粪便MT治疗CDI的成功率接近100%,几乎没有副作用(van Nood等人,2013年;Li等人,2016年)。这表明可以通过引入新的微生物来改变疾病表型。
微生物群相关疾病的另一个例子是肥胖症,它影响到全球超过三分之一的20岁以上人口。这种疾病是遗传、饮食、体育锻炼和其他几个因素共同作用的复杂特征的典型例子。旨在单独治疗这些因素的医疗程序显示成功率较低。然而,微生物移植被认为是一种潜在的解决方案,因为其治疗感染的成功率很高。尽管有几项研究发现肥胖者和瘦人之间的微生物门存在差异,但其相对比例尚未得到一致报告(Jayasinghe等人,2016),因此很难提出导致肥胖的特定门。从这个意义上讲,与肥胖相关的微生物变化可能与受试者依赖的微生物种群结构有关,例如某些物种或功能的相对丰度,而不是与特定门的存在或缺失有关(Walters等人,2014)。到今天为止,MT尚未被批准用于治疗肥胖的人体试验。然而,据报道,当瘦个体从超重者那里获得微生物群以治疗CD感染时,受者体重显著增加,这表明MT也可以改变人体体重(Alang和Kelly,2015)。
人类的机器翻译现在受到法律的高度管制,需要获得特别许可才能实施,因此大多数机器翻译的研究都是在小鼠身上进行的。具体来说,无菌(GF)小鼠通常用作微生物移植的受体。GF小鼠是被称为遗传生物的一大类动物的子集,包括所有体内具有特定和已知微生物群的动物,包括缺乏任何种类微生物的动物。GF小鼠已被证明是研究微生物移植的优秀模型,因为宿主的微生物群落以及许多其他变量(如遗传、饮食和环境)都是受控的。据观察,肥胖表型可通过GF小鼠的微生物移植传播(Turnbaugh等人,2006)。与从瘦供体获得MT的GF小鼠相比,从肥胖小鼠获得微生物群落的GF小鼠的体脂显著增加。尽管小鼠体内的微生物群落不同于人类,但已发现它们之间的肠道微生物组群有相似之处。一个例子是,与瘦受试者相比,肥胖受试者中类杆菌/厚壁菌的比例,这使得这些动物似乎是人类肠道研究的良好替代品。事实上,跨物种机器翻译已经在人类和小鼠之间取得了成功。将肥胖人类的粪便微生物群落移植到GF小鼠后,建立了人类微生物,并将其与肥胖表型一起跨代传播,这表明重要表型可以通过微生物群跨多代传播(Turnbaugh等人,2009b)。
除了这些水平机器翻译的例子外,还有自然和垂直的方式将微生物群落从父母传递给后代。例如,出生时的分娩方式对儿童微生物群的发育有重要影响(Dominguez-Bello等人,2010)。当对胎粪样本进行微生物分析时,观察到剖腹产婴儿的微生物成分与母亲皮肤中的微生物成分相似。相比之下,阴道出生婴儿的微生物群与母亲的阴道更相似。此外,剖宫产新生儿的微生物群与更高的CDI风险相关,并且与肥胖相关的类杆菌定植较低。与阴道出生的儿童相比,同一组儿童的肥胖率增加46%,患1型糖尿病的风险增加20%(Cardwell等人,2008年)。据推测,初始细菌在确定其他菌株的建立过程中起着重要作用(Fanaro等人,2007年),并促进肠道其他微生物群的正确建立。
母乳喂养是人类微生物垂直传播的另一种途径。已观察到配方奶粉喂养和母乳喂养婴儿之间的重要微生物差异。母乳喂养的新生儿经常表现出较高的双歧杆菌属丰度,这与健康有关,而配方奶粉喂养的新生儿的微生物数量较低。
母乳喂养VS配方奶粉喂养,对于微生物数量有影响。同时,顺差和剖腹产对于婴儿体内的微生物也有影响。
8. 我们微生物群落的遗传背景
人体不仅为其微生物提供了一个相对稳定和丰富的环境,而且影响其组成和行为。肠道微生物种类及其丰度不断受到肠上皮中抗菌肽释放的控制。众所周知,对于大多数人体部位,宿主遗传学与微生物组的建立和稳定性有关(Blekhman等人,2015)。例如,除了与疾病进展相关外,一些个体基因还对肠道微生物群的多样性和结构产生重大影响。LEP基因,也称为OB或leptin编码基因,就是一个很好的例子。这种特殊的激素作为一种细胞因子,可以控制食欲、能量消耗和其他改变微生物组成的代谢过程。LEP分泌与宿主中的脂肪量直接相关,这也与特定微生物的增殖有关。瘦素基因(ob/ob)被破坏的小鼠出现肠道失调。与正常小鼠相比,在这些动物中,类杆菌门的丰度较低(OB/OB)(Ley等人,2005),这种变化导致从饮食中获取能量的能力增加,因此增加了宿主的肥胖(Turnbaugh等人,2006)。有关直接影响微生物组组成的其他基因改变示例,请参见Spor等人(2011)。
在所有可能的人类遗传变异中,与免疫反应相关的基因中发生的变异对微生物组成的影响最大。与我们的免疫系统直接相关的基因发生突变,如影响免疫球蛋白、HLA或防御素的基因,可以改变我们与微生物、病原体或其他微生物的相互作用方式。GWAS提供了检测宿主遗传学和微生物组特征之间相关性的技术。然而,这些研究不是分析特定基因及其对微生物组的影响,而是研究人类遗传多样性及其与微生物物种异质性、相对分类群丰度及其功能的关系。通过研究近100名受试者的遗传和微生物组成,Blekhman等人发现基因组序列的相似性与微生物组的相似性之间存在正相关(Blekhan等人,2015)。事实上,在三分之二的受试身体部位中,宿主遗传学和微生物组分之间存在着实质性联系,特别是在免疫相关途径中。有趣的是,相关的人类基因组区域显示出高度分化,表明宿主的基因组适应特定的环境和微生物。
由于我们的基因影响我们的微生物组,反之亦然,人类相关微生物及其各自的丰度高度个性化的事实变得更容易理解。如今,研究样本中不太占优势的分类群,不仅可以让研究人员区分肥胖和瘦削个体,还可以检测样本来源的身体部位,甚至可以发现一组个体中宿主的身份(Ursell等人,2012)。每小时约有106个生物粒子从人体排放到周围环境。
因此,除了常见的粪便样本分析外,大多数人还可以通过其个人空气中释放的微生物进行识别(Meadow等人,2015年)。乍一看,这一结果与单个个体整个发育过程中微生物组的高度可变性不一致,因为微生物组的组成取决于饮食和其他环境因素。然而,正是对稀有微生物物种丰度的表征使得微生物组的个性化成为可能,即使个人生活在一个共享的环境中,这些物种也可能有所不同。这种高度个人化表明,人类遗传学和微生物组成之间存在着深刻的关系,支持这样一种观点,即个体内的表型相似性可能不仅来自人类基因,也来自微生物基因。类似地,最近的实验表明,种族和家庭关系也与整个微生物组成相关。通过分析来自马拉维和委内瑞拉美洲印第安人家庭的粪便样本,并将其与来自美国的样本进行比较,Yatsunenko等人发现,地理位置相近的个体之间的微生物多样性比生活在遥远地区的个体之间更为相似(Yatsuneko等人,2012)。此外,对人类细菌菌株水平和家族传播的早期研究表明,幽门螺杆菌的特定菌株仅在经常身体接触的人群之间共享。特别是,幽门螺杆菌分离株在美国的核心家庭成员中共享,但在南非没有。作者认为,这可能是因为在南非,没有任何亲属关系的人之间的身体接触更为常见,这与美国的社会规范形成了鲜明对比(Schwarz等人,2008年)。考虑到我们的个性化微生物群之间的微生物交换率很高,与陌生人的物理交互可能是我们微生物群配置的一个重要因素。这些结果表明,微生物群的组成不仅受我们的基因影响,还受我们的行为和周围环境的影响,使环境成分和基因成分更难分离。由于种族和血缘关系是遗传相似性的代表,如果不考虑微生物组多样性,则受微生物组影响的表型的遗传力计算可能会有很大偏差,因为微生物组成可能会受到这些常见习惯和行为的影响。因此,两个因种族而具有紧密遗传相似性但饮食习惯不同的个体可能表现出较低的表型相似性但较高的遗传相似性,这会影响所述表型遗传力的计算。
简而言之,相关个体的微生物组比非相关个体的更相似(Ursell等人,2012年)。微生物转移可能发生在身体亲密或生活在类似环境中的个体之间,例如家庭中的亲属。因此,如果表型的发展与微生物组有关,仅观察单个人类遗传多态性将无法揭示属于不同个体的微生物之间的遗传内容和功能的相似性。因此,表型方差和观察到的基因型方差之间将出现差距,在熟悉的聚类中测量的遗传力估计值较高,而在GWA中估计值较低。
9. GWAS和MWAS
关于我们的基因组对微生物群的影响程度,双胞胎研究是一个极好的信息来源。当个体的基因相同时,复杂的特征(如体重增加)可能更容易理解人类基因对微生物群的影响。众所周知,体重增加有重要的遗传成分,但也有很强的微生物影响。基因变异和运动、饮食等因素已被证明会影响体重增加。通过研究这种表型作为对双胞胎饮食的反应,双胞胎(同一对中)之间的表型相似性大约是双胞胎(不同对)之间的三倍,这表明个体之间的遗传相似性越强,表型变异越低(Bouchard等人,1990)。然而,这项研究仅着眼于人类的基因特性,假设它来自血缘关系,但没有着眼于微生物组的相似性。同卵双胞胎中的表型相似性可能来自这样一个事实,即他们之间的微生物组比其他双胞胎中的更相似(可能是由于类似的环境因素),并且观察到的体重增加的相似性不仅是由于人类细胞中的遗传变异,而且是由于微生物功能。例如,饮食偏见在双胞胎中具有高度相关性。与双卵双胞胎(DZ:“异卵双胞胎”)相比,单卵双胞胎(MZ:“同卵双胞胎”)之间的食物选择更为相似,并且在运动反应中观察到了类似的结果(Savard和Bouchard,1990)。尽管这些因素与微生物组之间的关系尚未完全确定,但众所周知,饮食和运动与体重增加直接相关,也会影响我们的微生物组成(Spor等人,2011;Kang等人,2014)。
遗传力研究通常使用MZ和DZ双胞胎,以更好地控制遗传和环境因素。在这些研究中,通常通过比较MZ和DZ对之间特定表型性状的相关水平来测量遗传力。具体地说,Falconer公式表明,性状的遗传力可以通过以下公式来衡量:
h 2 = 2 ∗ ( R M Z − R D Z ) h2=2*(RMZ−RDZ) h2=2∗(RMZ−RDZ)
上面是通过同卵双胞胎和异卵双胞胎的变异,来计算遗传力,这个遗传力是最准确的,是我们认为的真实的遗传力。
其中h2表示表型的遗传力,R表示每对双胞胎之间的相关性。因此,如果RMZ>RDZ,该性状将具有一定程度的遗传力,因为表型变异与遗传特性相关。对微生物群遗传力的早期研究发现,MZ双胞胎的微生物群之间的相似性比DZ双胞胎的更大(这意味着微生物群的RMZ>RDZ,Zoetendal等人,2001;Stewart等人,2005),这一发现最近得到了证实(Goodrich等人,2014,2016)。因此,如果将微生物群的组成视为另一个表型,则微生物群的RMZ>RDZ这一事实表明存在一定程度的遗传性。观察到几个微生物群可以跨代遗传,还发现了人类微生物基因关联,进一步证实了我们的基因含量影响我们的微生物群,我们的微生物组影响我们的表型的观点。因此,尽管许多环境因素在构建微生物组中起着重要作用,但遗传成分似乎也起着作用。值得注意的是,这些研究关注的是微生物特定分类群或门的遗传力,正如我们下面讨论的那样,可能不如关注其功能重要。
10. 通过MWAS重新审视遗传力
有争议的是,另一项双胞胎研究没有检测到微生物组的任何可遗传成分(Turnbaugh等人,2009a)。在本研究中,通过16S rRNA序列测量了每个双胞胎粪便微生物组的多样性。通过广泛使用的UniFrac距离测量MZ和DZ对之间的相似程度,结果表明RMZ和RDZ之间没有显著差异,表明微生物组缺乏遗传力。在所有样本中,丰度超过0.5%时,未发现系统类型,这进一步证实了他们的结论。事实上,在筛选健康人群的共享微生物类群时,没有观察到特定类群在个体中普遍存在(Huttenhower等人,2012年)。这些结果可能表明微生物组的遗传力较弱或是间接的。然而,重要的是要记住,UniFrac距离并不能解释微生物组的功能,即使在系统发育密切的个体之间,功能也可能不同。UniFrac距离是一种β-多样性度量(生态系统之间的多样性),其中,如果两个个体的微生物群落成员在系统发育上更接近,则两个人的微生物组将更相似。在这个意义上,微生物组的功能被认为是系统发育相似性的结果。我们现在知道,亲缘关系并不一定意味着类似的功能。事实上,一种分析双胞胎中微生物群落基因含量的宏基因组方法发现了许多在他们之间共享的基因。然而,他们没有使用这些数据计算微生物组的遗传力。尽管如此,这些结果允许在基因水平而不是物种水平上鉴定“核心微生物群”。因此,我们有理由认为,正是我们的微生物的功能被强烈地选择用于。微生物组的功能不一定与特定分类群相关,这在微生物组疾病关联中经常被假设。如前所述,个体间微生物组的分类多样性非常高,而代谢途径保持相对稳定(Huttenhower等人,2012)。即使在同一属内,不同的菌株也可能有非常不同的代谢途径(Huttenhower等人,2012年)。例如,当在200多个个体中测量链球菌的携带情况时,每种链球菌的丰度在他们之间差异很大。菌株水平的基因组变异和基因丢失非常高,表明单个菌株的丰度与不同代谢途径之间存在联系。这些事件与宿主特异性基因组变异有关,这间接意味着个体内链球菌物种的功能多样性将部分取决于宿主的遗传学,而不是取决于这种细菌的特定菌株。这表明,与特定菌株相反,任何微生物相关性状的遗传力最有可能发生在功能途径方面。虽然是间接的,但迄今为止提出的证据有力地表明,将我们微生物的基因型和功能多样性纳入表型关联研究,从而纳入遗传力估计中是重要的。在下一节中,我们指出了一些将微生物组的功能多样性纳入表型预测的方法。
当试图通过纳入微生物数据来估计表型的遗传力时,应考虑几个重要因素。首先,尽管微生物的组成可能在一定程度上取决于我们的基因,但微生物基因不是我们的。如果我们使用遗传力的定义,即由人类遗传变异解释的总表型变异的比例,那么微生物将被视为我们遗传学的外部因素。然而,如果我们认为自己是一个生态系统或全生命体,微生物及其基因是其中的重要组成部分,正如我们所讨论的,它们能够影响我们的表型。因此,只有当我们了解表型的遗传程度,并缩小与家族研究估计的差距时,才可以自然地将其基因包括在内。其次,很明显,微生物的组成强烈依赖于环境。由于遗传力与遗传变异有关,而与环境因素无关,因此在包括微生物遗传学时,必须特别注意将环境因素与遗传因素分开。微生物组似乎模糊了作用于表型的遗传力和环境力之间的界限,因为微生物组的遗传成分比我们自己的更灵活,并且可以根据外部因素(如抗生素摄入、感染、饮食变化等)而变化。这种灵活性使分析更加困难,因为分类群可以在短时间内发生变化。然而,基因含量已被证明是一种在时间上和在个体之间更为一致的测量方法,以至于可以用功能术语定义核心微生物组。这在确定影响表型发育和遗传力的微生物遗传因素时非常有用。整合来自GWAS和MWAS的数据并不简单。GWAS数据是分类的,每个SNP都有一组离散的值。值可以是0、1或2,这取决于主要(0)或次要等位基因(2)的基因型是纯合的,还是杂合的(1)。然而,MWAS处理的是连续金额。事实上,分类群丰度不仅是连续的,而且是根据相对丰度给出的。因此,在给出任何有价值的解释之前,必须仔细地进行规范化。微生物功能数据也根据丰度给出,因此每个基因或基因簇都有相应的丰度,其与表型的相关性可以评估。将微生物数据纳入GWAS以估计性状遗传力的一种可能性是,像数据挖掘技术中通常所做的那样,将连续变量离散化到箱子中。这种离散化可能并不简单,因为它可能取决于总体中的观察值。然而,一旦完成这一步骤,就可以使用几种方法来估计与表型的相关性(Lee等人,2011;Bloom等人,2013)。如果设计得当,双胞胎研究对控制环境和人类遗传因素非常有用。例如,在婴儿期,当饮食和行为习惯几乎相同时,对双胞胎的微生物组群进行分析的后续研究可以深入了解其组成在以后的生活中如何改变,而这些环境因素不再相似。这可能有助于解释环境对微生物组成的影响程度,以及这种差异在多大程度上与肥胖、糖尿病或其他几种胃肠道疾病的出现相关。最后,已知微生物组的组成和功能随年龄、性别和种族而变化,因此在研究设计中考虑的其他重要因素应涉及人口分层。使用GWAS和MWAS计算性状的遗传力肯定不是一项简单的任务,需要开发新的统计技术。然而,我们认为这是一项值得做的工作,因为这些方法无疑将在正确评估微生物组在生理和进化中的作用方面发挥重要作用,在被视为生态系统的新兴生物体范例中发挥重要作用。
基于GWAS研究,有几种技术可用于计算表型的遗传力(Zaitlen和Kraft,2012)。如下所示,其中一些技术可以用于包括微生物宏基因组数据,以估计特定表型的狭义遗传力。数学上,狭义遗传力定义为
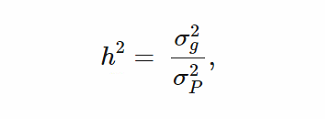
上面是,传统遗传力的计算公式
其中σ2g是加性遗传方差,σ2P是表型方差,定义为:
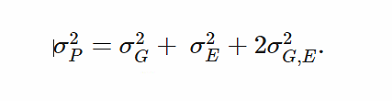
当有基因与环境互作时的遗传力公司
在最后一个表达式中,σ2G是总遗传方差,σ2E是环境方差,而σ2G,E是遗传成分和环境成分之间的协方差,这通常被假设为不存在。在加性模型中,第j个个体(yj)的表型由其遗传变异的线性效应总和定义:
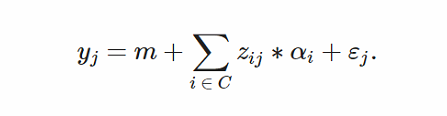
在这个方程中,m是从线性回归中导出的常数,zij是如下所示的归一化基因型(代表MWA中的离散丰度),C是基因型SNP的集合(MWAS中的基因或基因簇),αi是每个变量的效应大小,εj是环境贡献。归一化基因型zij计算如下

当该公式用于使用从GWAS获得的人类基因型计算表型的遗传力时,gji代表第j个个体的第i个基因型。
这个是根据MVA的离散丰度计算的类似G矩阵的Z矩阵
gji公司∈ {0,1,2},取决于第j个个体在第i个SNP中主等位基因(0)的纯合、杂合(1)或次要等位基因的纯合(2)。这里pi是第i个SNP中次要等位基因的概率。在GWAS中,性状的遗传力计算为每个标准化基因型的平方效应大小之和(Zaitlen和Kraft,2012):

效应大小通常通过Cohen的D参数计算,该参数衡量特定SNP对表型的影响有多大。
然而,新的宏基因组技术允许我们使用特定微生物功能/基因的丰度而不是基因型来计算效应大小。这是因为MWA报告了微生物组中所有基因的丰度,而不管它们属于哪个特定的生物体。这些丰度必须离散为群体中每个基因的k值(或类别)。在对每个个体的每个微生物基因/功能进行分类后,效应大小可以计算为:
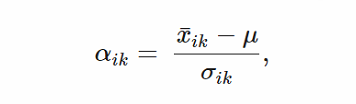
其中,x´ik是属于基因i的k类的所有个体的平均表型值,μ是群体的平均表型价值,σik是表型标准差。需要注意的是,根据标准偏差σik的选择,存在几种效应大小的定义。例如,科恩的D使用
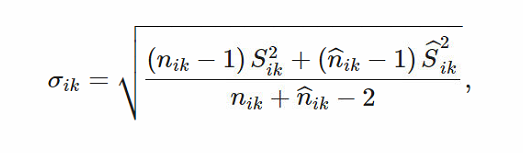
其中nik是属于第i个基因第k个类别的群体中的个体数量,Sik是该数量的标准偏差,而nˆik和Ŝik是不属于第i基因第k类的个体的相应数量。
通过计算每个微生物基因中每个类别的效应大小,我们现在可以计算微生物基因对相关性状遗传力的线性贡献。由于群体表型方差归一化为1,σ2P=σ2g+σ2E=1,因此可以直接获得狭义遗传力的微生物贡献:

其中σ2g是使用基因丰度计算的所有α2ik的总和。由于h2MWAS估计遗传微生物成分对表型遗传力的线性贡献,因此可以直接将其添加到通过GWAS计算的遗传力中。我们提出,表型的总狭义遗传力必须计算为
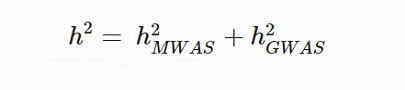
整体而言,所有的遗传力,应该是MWAS和GWAS的遗传力之和。这个GWAS的遗传力不是显著性SNP的遗传力,而应该是所有SNP的SNP遗传力。
11. 结论和展望
为了总结本文提出的观点,我们将体重视为一个特征的例子,其中人类和微生物遗传学是理解缺失遗传力问题的关键因素。从双胞胎和家庭研究中首次估计出的体重指数(BMI)的遗传力约为45%。最近的研究发现遗传力在50%到90%之间(Elks等人,2012年),但当使用GWAS搜索与体重指数相关的单核苷酸多态性时,发现的多态性只能解释2%的表型变异。鉴于两个测量值之间的巨大差距,这一结果清楚地说明了缺失遗传性问题。肥胖无疑是一种涉及遗传和环境因素的多因素疾病。然而,母亲肥胖和糖尿病始终是儿童肥胖最有力的预测因素(Redsell等人,2013年)。研究表明,母体西方饮食可促进小鼠的脂肪毒性和脂肪肝疾病。此外,它减少了肠道微生物群的多样性,这些微生物群以及脂肪毒性和脂肪肝在后代中持续存在,即使在改变为更健康的饮食后。请注意,对于我们的假设尤其重要,即后代也缺乏微生物多样性(Brumbaugh和Friedman,2014)。从这些和其他研究(其中一些之前已经讨论过)中,我们得出结论,此类肥胖相关治疗的遗传不能仅归因于小鼠基因的传递。这反过来表明,由于暴露于超重父母的肥胖相关微生物群(母乳喂养、子宫定植或分娩方式)而导致的初始失衡可能会产生挥之不去的后果,并可能导致后代肥胖。
MWAS和16S rRNA测序表明,肥胖和瘦削个体在物种多样性和基因计数方面存在明显差异(Le Chatelier等人,2013年)。这些观察结果强烈表明,当谈到体重指数的表型变化时,微生物成分在决定体重增加方面起着重要作用。因此,在家族研究中对体重增加遗传力的估计可能严重高估了人类遗传学的作用(遗传力高达90%,而在GWAS中为2%),并且根据我们的假设,这种遗传力可能来自这些家族中共享的微生物的线性遗传效应。我们认为,如果考虑到微生物组成和功能之间的相似性,大多数BMI和其他人类表型的遗传力估计将显著提高。
为了使本文内容完整,我们首先回顾了微生物群在生理学中的作用。然后,我们提出了支持以下假设的证据:为了提高我们对表型遗传力的估计,必须考虑微生物组。我们的思路如下。首先,GWAS未能找到从双胞胎和其他家族研究中获得的报告遗传力。尽管表观遗传学和上位性被认为是表型变异的替代来源,但对于遗传力的产生机制或载体尚未达成共识。此外,迄今为止,还没有任何研究考虑到微生物组及其功能和遗传潜力,试图解决缺失遗传力问题。正如它影响的大量表型所证明的那样,考虑到这一巨大的变异来源至关重要。这方面的例子包括免疫系统发育、肥胖、糖尿病、克罗恩病、肠易激综合征、癌症和神经系统疾病。我们在本文中还强调了这样一个事实,即我们的身体可以对我们的微生物群的结构和组成产生强大的影响。众所周知,饮食调整和抗生素的使用会快速、剧烈、有时甚至永久地改变我们的微生物群。此外,我们身体释放的代谢物可以调节微生物群落的行为,这是微生物群落与人类生理学之间生态相互联系的指标。就这些微生物的遗传而言,已经证明,个体之间同时发生水平和垂直传播,并且这些传播能够影响受体的表型,例如使其更容易受到疾病的影响或抵抗疾病。在个体之间成功传播的特定微生物的性质也可能取决于宿主的遗传。
但是,由于亲属关系或种族不仅是遗传身份的代表,而且也是行为和饮食(可能影响微生物组成)的代表,基于不考虑微生物遗传变异的家庭研究的遗传力估计导致了这样一种看法,即表型变异仅是由于人类遗传相似性引起的。由于微生物组分与种族或亲属关系之间的相关性可能是由共同的环境、相似的饮食和其他习惯引起的,因此必须对单卵和双卵双胞胎进行研究,以允许对这些变量进行一定程度的控制。一些研究表明,MZ双胞胎的微生物群之间的相似性仅略高于DZ双胞胎,而其他研究表明,没有遗传特定的分类群,微生物群的差异非常小。尽管如此,当考虑到这些微生物群的功能时,确实发现了许多共享基因,允许在功能水平而不是物种水平上识别“核心微生物群”。
我们在本文中提出,在评估任何人类特征的遗传力时,最重要的是在研究组之间寻找共享的微生物组功能,而不是特定的分类群。实现这一点的有希望的方法包括结合GWAS和MWA、数据挖掘算法(如决策树)、贝叶斯推理和监督分类等。这种个体遗传多样性及其微生物组的整合很可能会导致对许多重要表型的遗传力进行更准确的计算,不仅是在人类中,而且在牲畜和植物中。