《Correcting for batch effects in case-control microbiome studies》
- 文章地址:https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1006102
- DOI:https://doi.org/10.1371/journal.pcbi.1006102
- 期刊:PLoS Computational Biology
- 2022年影响因子/中科院分区:4.779/中科院1区
- 发布时间:2019年1月24日
- 校正病例对照微生物组研究中的批次效应
- github:https://github.com/seangibbons/percentile_normalization
- 摘要:
- 介绍
- 方法
- 数据
- 数据预处理
- 百分位标准化
- combat
- limma
- 数据分析
- 计算实验
- 结果
- 批次效应在 OTU-level 上解决
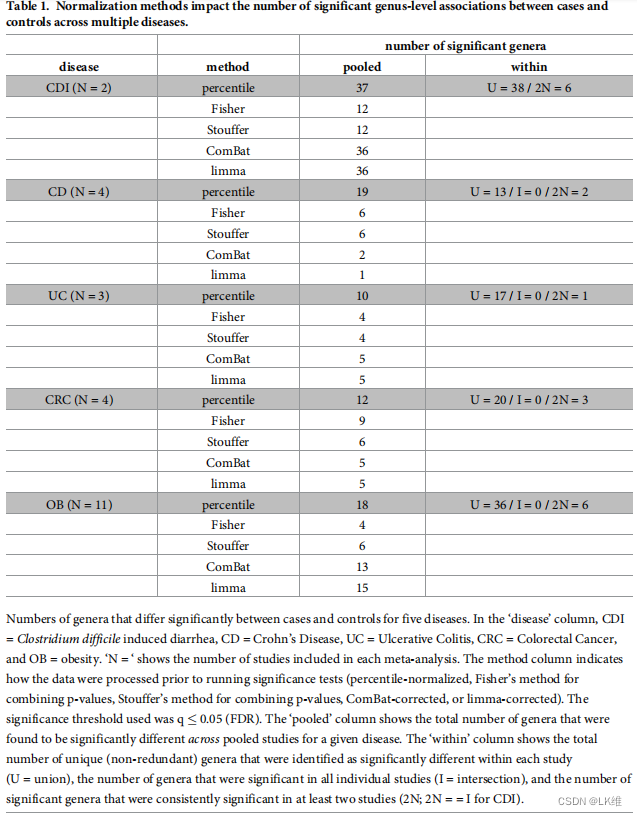
- 批次效应在属级跨多种疾病上的解决
- 讨论
文章地址:https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1006102
DOI:https://doi.org/10.1371/journal.pcbi.1006102
期刊:PLoS Computational Biology
2022年影响因子/中科院分区:4.779/中科院1区
发布时间:2019年1月24日
校正病例对照微生物组研究中的批次效应
github:https://github.com/seangibbons/percentile_normalization
摘要:
高通量数据生成平台,如质谱、微阵列和第二代测序,由于试剂、设备、协议或人员的运行变化,容易受到批处理影响。目前(文章投稿于2018),批量校正方法并不常用于微生物组测序数据集。在本文中,我们比较了不同的批校正方法应用于微生物组病例对照研究。我们引入了一种无模型的归一化程序。在研究中,在跨研究样本汇集数据之前,将样本的特征转换为对照样本中等效特征的百分位数。我们引入了一种无模型的归一化程序,在研究中,在跨研究样本汇集数据之前,将样本的特征转换为对照样本中等效特征的百分位数。我们将研究这种百分位数归一化方法与结合独立p值以及limma和ComBat的传统元分析方法之间的比较。这些方法广泛用于为RNA微阵列数据开发的批量校正。总的来说,我们表明百分位归一化是一种简单的、非参数的方法,提高病例与对照组区分的敏感性。
介绍
通过质谱、第二代测序或微阵列等高通量方法产生的数据对实验和计算处理很敏感。这种敏感性使得在一个独立的实验运行之间产生了“批处理效应”。当不同的研究小组坚持相同的方法时,这些影响可能会由于硬件、试剂或人员的轻微差异而产生。因此,对不同研究之间的未校正数据进行直接的、定量的比较是不合适的。
几种可以减少RNA微阵列数据中的批处理效应的工具已经被开发出来。例如,替代变量分析(SVA)估计了一组推断变量(特征向量),以解释与假定的批处理效应相关的方差。然后,将这些推断出的变量纳入一个线性模型,以纠正下游的显著性检验。limma包采用了类似的线性校正来解释批处理效应。SVA和limma是线性批校正方法家族的一部分,它们使用不同种类的因子分析、奇异值分解或回归。迄今为止最依赖的方法被称为ComBat,它使用贝叶斯方法来估计一批中每个特征的位置和尺度参数。当批效应不与真正的生物效应合并时,所有这些模型都是最有效的。此外,大多数批校正方法都做出一定的参数假设。
不幸的是,通常对许多类型的组学数据都有效的模型可能不适用于微生物组数据集。在微生物组研究中,批效应通常会弥散的并与生物信号合并。微生物组领域也在努力寻找细菌丰度分布和处理零样本。对于微生物组测序研究中的低生物量样本尤其如此,比如从建筑环境中提取的样本,种群采样不足,生物信号相对较弱,批效应可能相当大。解决这个问题的一种方法是计算给定批内的统计数据,然后使用经典的元分析技术组合p值来比较跨批次的重要特征,如Fisher和斯托夫的方法。这些meta分析技术对独立研究的批处理效应是稳健的,但与直接汇集跨研究项目的数据相比,它的统计能力和检测细微差异的能力更低。
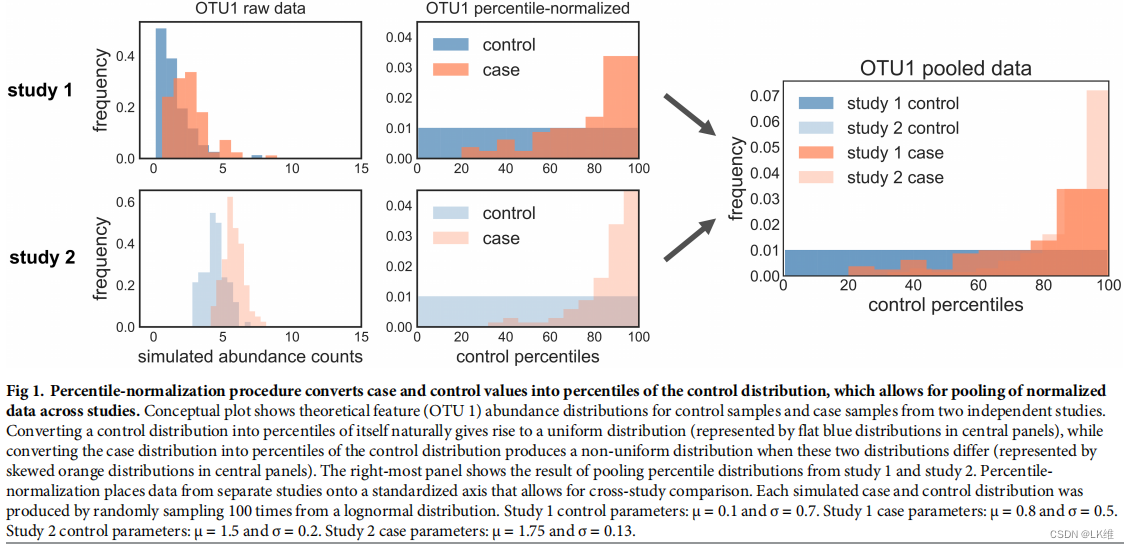
在这里,我们描述了一个无模型的数据归一化程序,用于控制病例-对照微生物组研究中的批处理效应,从而能够在各个研究中汇集数据。病例对照研究包括一个内置的对照样本群体(如健康受试者),可用于使病例样本(如患病受试者)正常化。对于每个特征(即细菌分类单元),病例丰度分布可以转换为等效控制丰度分布的百分位数(Fig 1).。病例样本中出现的研究特定批效应也将出现在对照样本中,通过将病例数据转换为对照分布的百分位数,这些影响得到减轻。在转换为研究内对照的百分位数后,来自具有相似病例对照定义的多个研究的周样本可以更适当地合并进行统计检验(Fig 1)。我们表明,这种方法有效地控制了微生物组病例对照研究中的批效应,我们将这种方法与汇集战斗或边缘校正数据,以及Fisher和斯托弗的结合独立p值的方法进行了比较。我们将这种方法与ComBat或者limma数据进行比较,以及与Fisher和斯托弗的组合独立p值的方法进行比较。
方法
数据
数据来源:MicrobiomeHD database 16S gene (V4)
我们的分析重点集中在5种疾病的研究上:
- 结直肠癌(CRC)
- 克罗恩病(CD)
- 溃疡性结肠炎(UC)
- 肥胖(OB)
- 和艰难梭菌诱导腹泻(CDI)
数据预处理
OTU-level
方法:Duvallet et al.(2017)所述,对每个V4数据集进行了质量过滤和长度裁剪,并将这些原始的、裁剪过的FASTQ文件连接到一个文件中。删除出现不超过20次序列,并将剩余的reads以97%的相似性进行聚类。使用RDP分类器分配这些OTUs分类标识符,截断值为0.5。
genus-level
处理方法同Duvallet C (2017). 对每个研究的OTU表将每个样本除以其总reads数,转换为相对丰度,并将具有相同属级注释的所有OTUs相加,分解为属级。
在排序空间中绘制数据
Bray-Curtis distances使用Scikit-learn从相对丰度数据中计算出。
Non-metric multidimensional scaling (NMDS)两个轴的坐标的计算是基于Bray-Curtis distances使用Scikit-learn
百分位标准化
在这个过程中,控制特征分布对自身进行百分位归一化(导致在0到100之间的均匀分布),情况特征分布是转换为其等效控制特性的百分位数。将我们的对照视为零假设是基于这样一种想法,即健康患者应该在不同的数据集上被视为相似的患者,尽管我们理解它们会由于生物和技术批效应而有所不同统计法转换为百分位数。为了避免由于存在多个零而导致的秩堆积,我们用0.0到10−9之间均匀分布的伪相对丰度替换零。由于零替换步骤,在用不同的随机抽取重新分析时,p值可能会发生轻微的变化,这可能导致非常接近显著性阈值的特征的显著性损失或获得。在每项研究中,每个OTU或属的对照分布被转换为自身的百分位数,病例分布被转换为相应对照分布的百分位数。我们已经编写了一个python脚本,它给定一个OTU表、一个用例样本id列表执行百分比规范化,以及一个控制示例id列表作为输入 (https://github.com/seangibbons/percentile_normalization),还有一个可用于运行百分位数规范化的QIIME 2(https://qiime2.org)插件(https://github.com/cduvallet/q2-perc-norm)。
combat
在运行ComBat之前,对CRC分析中的相对丰度(CRC分析中的OTUs崩溃到属级或在属级分析中的OTUs)进行对数转换,添加最小频率一半的伪相对丰度(跨越整个特征表)来替换零。然后在下游分析之前,combat校正数据从对数空间转换回来(即指数转换)。
limma
如上所述,将相对丰度(零替换为伪相对丰度,等于整个特征表中最小频率的一半)进行对数转换,然后使用removeBatchEffect 功能(默认设置)拟合一个线性模型来减去批处理效应。然后,在进行下游分析之前,将经过limma校正后的数据从对数空间转换回来(即指数转换)。
数据分析
为了计算统计意义,我们将我们的统计检验限制在至少三分之一的对照组或三分之一的病例样本中发生的OTUs/属,以减少我们的多重检验校正惩罚。我们使用Wilcoxon秩和检验,如在SciPy中实现的v0.19.0(sicipy.stats.ranksums),以确定独立样本组之间的显著差异。Wilcoxon测试是在研究内部或跨研究中进行的。为了计算跨研究的统计数据,将来自同一疾病的多个研究的归一化病例样本和对照样本合并到同一个OTU表中。此后,组合数据集被称为“池化”。p值使用本杰姆尼-霍赫伯格错误发现率(FDR)程序进行了多次检验校正,正如在StatsModels v 0.8.0 (statsmodels.
sandbox.stats.multicomp.multipletests)
计算实验
在计算显著差异之前,我们使用OTU-level数据进行了一次计算实验,该数据模拟了来自不同数据集的对照样本的池化。将一项研究的健康样本与另一项研究的健康样本以不同比例混合,然后计算病例和对照组之间OTU频率的显著差异。病例组和对照组各抽样40例。对照样本被从另一项研究中随机选择的沿着分数梯度的样本(从另一项研究中选择的0-100%的对照样本)所取代。我们使用Wilcoxon秩和检验计算了病例组和对照组之间的显著性差异,并应用了FDR校正。q值为<=0.05的OTUs被认为是显著性的。重复进行滴定实验20次,并取平均值。
结果
与滴定实验类似,我们通过OTU-level分析批校正方法如何影响假阳性率,随机选择40份对照样本(2016)研究作为人工对照和40份研究作为人工数据类型(通过20次迭代),每种数据类型(即原始、边缘化、边缘校正和作战校正)。然后,我们计算了上面概述的这些人工“病例”组和“对照组”之间的显著差异,以生成每个OTU的p值。
批次效应在 OTU-level 上解决
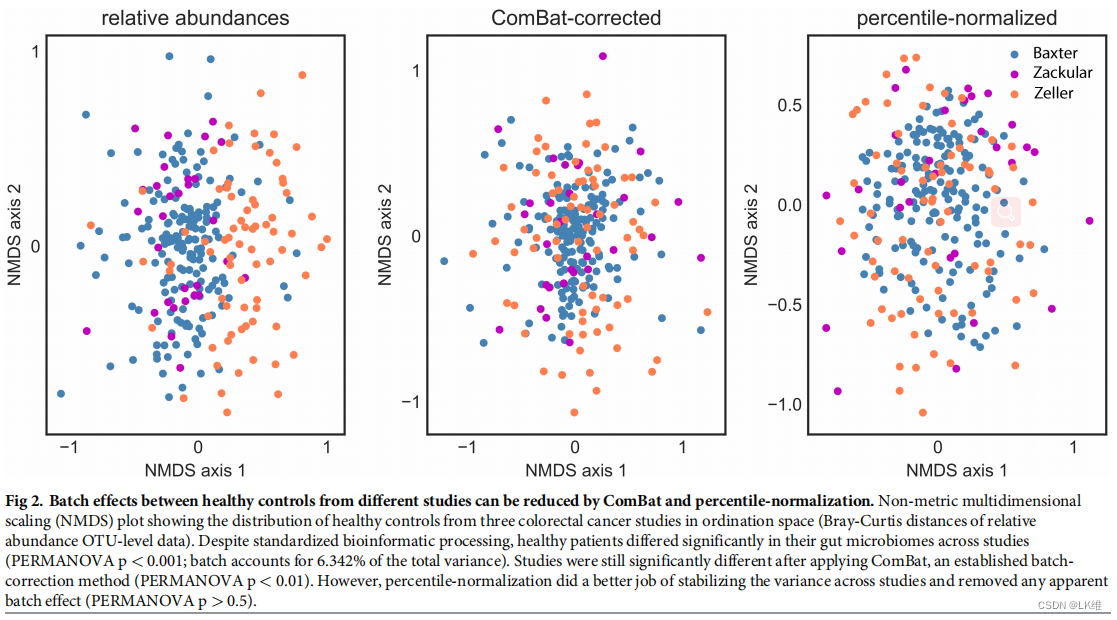
上图经百分位处理后,分散的更均匀。
图3 将一项研究的健康样本与另一项研究的健康样本以不同比例混合,然后计算病例和对照组之间OTU频率的显著差异
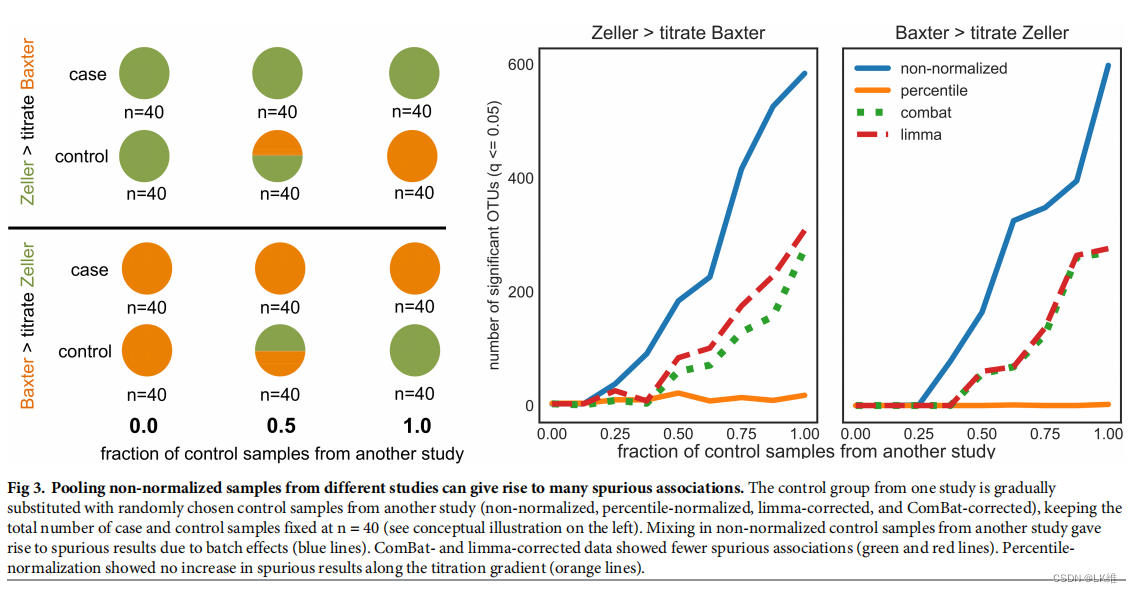
在control中增加其他样本的比例(逐渐增加),无批次处理的样本检测出来的重要OTUs数量增加。
而使用百分数方法检测出的量始终稳定。
批次效应在属级跨多种疾病上的解决
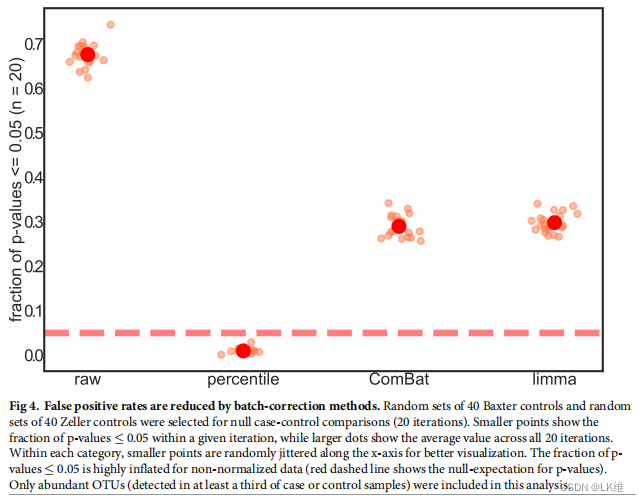
图4 显示使用百分数,假阳率更低
图5 在没有批处理效应的情况下,跨同一疾病的数据集汇集数据应能提高检测显著的交叉研究相关性的敏感性。
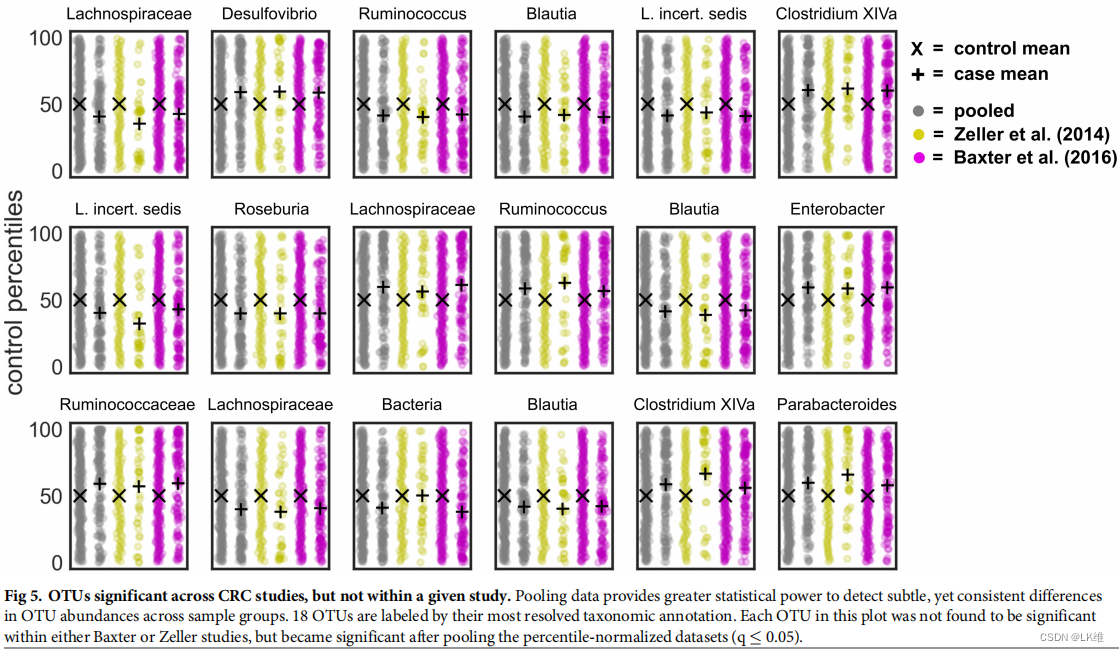
可以发现更多显著。
讨论
当使用高通量数据生成平台时,批处理效应是不可避免的。RNA微阵列社区一直积极开发处理这些影响的工具。然而,当批效应与生物信号混淆或参数假设不适用时,这些工具并不那么有效,这在微生物组病例对照研究中经常如此。因此,需要无模型的方法来纠正跨微生物组数据集的批处理效应。幸运的是,病例对照研究可以通过其自己的对照样本进行内部标准化。病例样本中任何特定于研究的批效应都将出现在对照样本中,通过将病例数据转换为对照分布的百分位数,这些影响在不做参数假设的情况下被减弱。
当对一项研究的病例进行测试时,, limma-corrected、ComBat-corrected数据,而不是来自另一项研究的对照数据,迅速产生了大量的虚假结果。此外,当对来自不同批次的对照人群进行比较时,非归一化数据比批量校正数据产生了更多的假阳性数量。我们的百分位数归一化方法在控制假阳性方面比limma和ComBat更有效,特别是在低丰度类群存在的情况下。
因为汇集数据集增加了统计能力,所以即使在缺乏合适的批处理校正方法的情况下,也很容易汇集这些数据。因此,在微生物组领域中,汇集来自不同批次的非归一化数据已经成为一种常见的做法。在本文中,我们演示了为什么这种做法是非常不可取的。汇集来自多个研究的批量校正数据,我们可以检测到在给定研究中没有发现的显著差异,同时消除了在不同研究中较弱或不一致的关联。百分位数标准化结果通常会发现其他标准化方法所遗漏的病例和对照组之间的显著差异。对于CDI,百分位归一化结果与其他批校正方法确定的显著命中数量大致相同,这可能是由于与腹泻相关的生物信号非常强。如果生物信号是强大的,结果应该对所使用的分析类型是稳健的。对于UC和CD研究(IBD),百分位数归一化确定了几个limma和ComBat没有的重要属。来自IBD的limma和ComBat校正数据的显著命中数量的减少可能是由于这些研究中的异质性对照队列(即健康患者与非IBD患者),这可能平滑了炎症相关信号。这一结果强调了对不同研究中的病例和对照队列有一致定义的重要性。我们比较了百分位数归一化和池化与Fisher和斯托弗的组合独立p值的方法。斯托弗的方法与Fisher的方法相似,但包含了基于研究中样本数量的每个p值的权重。百分位归一化与斯托弗和费舍尔的方法相比,我们始终能识别出更多的显著命中点,这证实了汇集数据增加了敏感性(即减少假定的假阴性)。结合来自独立研究的p值的方法是相当稳健的,可能应该被认为是一种安全的替代汇集(即较低的假阳性机会)。然而,在我们的肥胖分析中,费舍尔和斯托弗的方法认为莫吉细菌是重要的,尽管它在各个研究中明显不一致。
总之,我们提出了一种稳健的、无模型的程序,将微生物组病例-对照数据集中的每个特征转换为其控制分布的百分位数(图1)。应用这种方法的主要条件是:1)每批必须有相当数量的控制样本(即控制分布的密度限制了病例样本的分辨率),2)病例和对照人群(即“健康”或“患病”组的相同定义)。鉴于这些警告,百分位标准化特征可以在各个研究中合并,进行单变量统计检验(无论研究人员喜欢哪种检验——理想情况下是非参数的),从而缓解批效应问题。这种无模型程序也可以应用于其他类型的定义内部控制的组学数据集。我们发现,这个程序允许我们识别病例和对照之间的差异,这些差异往往被更保守的元分析技术所遗漏。为微阵列数据中开发的批校正方法,如limma和ComBat,可以部分减少微生物组研究中的批效应(图2-4),但如果批效应不是独立于生物信号或这些模型的参数假设是无效的,似乎掩盖了真实的模式。我们建议像limma和ComBat这样的方法对于缺乏病例组和对照组的研究是有用的。然而,当研究一致地定义了内部控制时,百分位数标准化应该是首选的批校正方法。未来的工作应集中于开发专门用于微生物组数据集的批量校正的参数模型,这可能进一步提高敏感性,以检测不同研究中细微的生物学差异。