实验篇——多序列比对,构树
文章目录
- 前言
- 一、名词解释
- 二、实操
- 1. 文件准备
- 2. 多序列比对
- 3. 对比对序列修剪
- 4. 构建进化树
- 5. 可视化进化树
- 总结
前言
系统发育树构建的软件大致有如下几种策略:从最简单的UPGMA法,到邻接法,最大简约法,再到最大似然法,以及贝叶斯法。其中最大似然法和贝叶斯算法是比较常用的构建进化树的方法。
而本文主要讲述的是基于最大似然法的构树。
一、名词解释
进化树是用于表示生物物种之间的进化关系的图形模型。它是一种类似于家谱图的树状图,通过分支的形式展示了不同物种之间的共同祖先和后代关系。
在进化树中,每个分支代表一个物种或群组或同一物种的样本、基因等单元,而节点则代表它们的共同祖先。树的顶端表示最近的共同祖先,而树的底端表示最近的后代。分支的长度通常代表了物种的进化时间,即时间越长,分支长度越长。
二、实操
1. 文件准备
通常是 fasta 格式的序列文件,可以包含 DNA 或氨基酸序列。如果是多个文件,先用 cat 将其合并成一个文件 seqs.fa
2. 多序列比对
使用muscle软件
#软件安装
mamba create -n wht_env3 muscle#比对
muscle -super5 /home/wuyao.pep -output /home/wuyao.afa
比对完成:
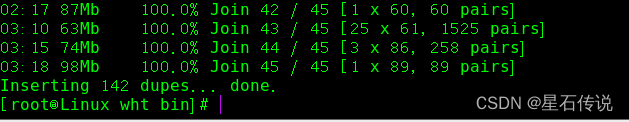
注:
下载的不同的muscle软件版本的使用命令可能不同,具体的命令使用可以先查看该软件的信息,如我下载的该软件:
3. 对比对序列修剪
使用trimal软件,提高构建进化树的准确性
#软件安装
mamba create -n wht_env4 trimal
#查看软件
./trimalsed -i 's/\*//g' /home/wuyao.afa
#软件使用
./trimal -in /home/seqs.afa -out seqs_trimmed.afa1 -automated1
4. 构建进化树
使用iqtree软件,进行最大似然法的进化树构建
#软件安装
mamba create -n wht_env1 iqtree#软件查看
./iqtree2#使用
./iqtree2 -s 
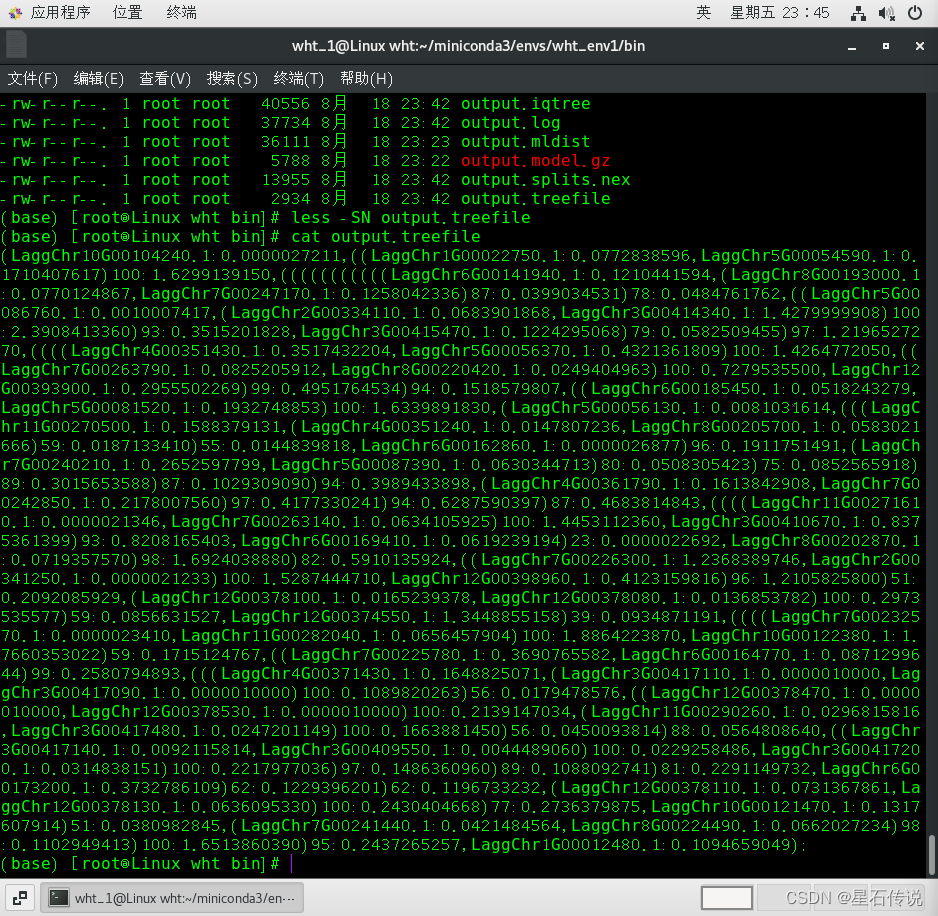
用 iqtree 构建输入文件 seqs_trimmed.afa1 的进化树,并输出为 output.treefile,查看此文件:
5. 可视化进化树
使用可视化软件,如Figtree,展示进化树(查看输出的进化树文件 output.treefile)
下载地址:
https://github.com/rambaut/figtree/releases
(注意:需要Java运行环境来运行Figtree,要提前下载好java)
打开软件:
导入树文件(output.treefile)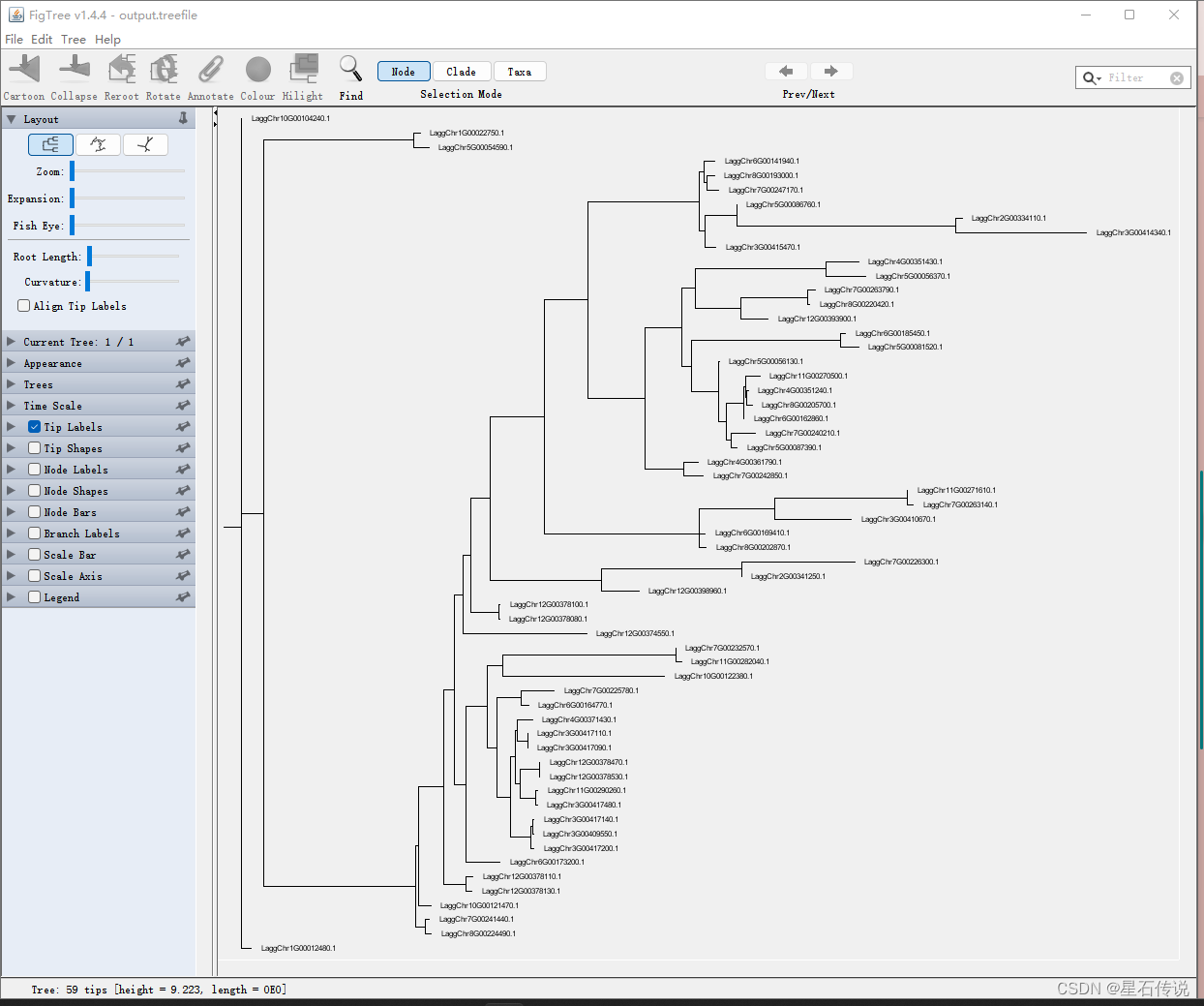
在左侧栏中可以对进化树设置,进行美化
总结
本章主要简述了在Linux中通过muscle软件进行多序列比对,然后经过trimal软件进行修剪,以提高构树的准确性,再然后则是使用iqtree软件得到树文件。最后将得到的树文件导入figtree软件中可视化,并且美化进化树。
最是人间留不住,朱颜辞镜花辞树
–2023-8-17 实验篇

![【傅里叶级数与傅里叶变换】数学推导——3、[Part4:傅里叶级数的复数形式] + [Part5:从傅里叶级数推导傅里叶变换] + 总结](https://img-blog.csdnimg.cn/77e1ce52f4ea477b86155158a95cfa46.png)