论文解析-moETM
- 参考
- 亮点
- 动机
- 发展现状
- 现存问题
- 功能
- 方法
- Encoder改进
- Decoder改进
- 评价指标
- 生物保守性
- 批次效应移除
- 实验设置
- 结果
- 多组学数据整合
- cell-topic mixture可解释性
- 组学翻译性能评估
- RNA转录本、表面蛋白、染色质可及域调控关系研究
- 1. 验证同一主题下,top gene可以映射到top protein
- 过程
- 结果
- 2. 跨主题验证gene-protein、peak-gene的调控关系
- 过程
- 结果
- 3. 验证重构的gene-protein、peak-gene更能反映组学相关性
- 联合peak-gene分析细胞类型特异性通路和调控机制
- 分析细胞类型特异性通路
- 分析细胞类型特异性motif
- 分析细胞类型特异性通路联合motif
- 补充
- 基因(蛋白)集富集过程
- 基因与翻译蛋白表达负相关的可能原因
- 寻找细胞标志物的资源--CellMarker数据库
- 寻找TF对应靶基因的资源--ENCODE Transcription Factor Targets
参考
Zhou, M. et al. Single-cell multi-omics topic embedding reveals cell-type-specific and COVID-19 severity-related immune signatures. Cell Reports Methods 3, 100563 (2023).

亮点
动机
从单细胞多组学数据探究生物模式的现存困难:
- 相比单组学技术,多组学技术通量更低,数据包含的细胞数量较少
- 不同组学结合之后特征维度更高,例如把scRNA-seq和scATAC-seq结合到一起
- 多组学数据噪声更多(解决方式:概率模型)
- 批处理效应
- 多组学测序技术成本更高(解决方式:用模型预测缺失的组学数据)
发展现状
| 方法 | 文献 |
|---|---|
| SMILE | Xu, Y., Das, P., and McCord, R.P. (2022). Smile: mutual information learning for integration of single-cell omics data. Bioinformatics 38, 476–486. |
| totalVI | Gayoso, A., Steier, Z., Lopez, R., Regier, J., Nazor, K.L., Streets, A., and Yosef, N. (2021). Joint probabilistic modeling of single-cell multi-omic data with totalvi. Nat. Methods 18, 272–282. |
| multiVI | Ashuach, T., Gabitto, M.I., Jordan, M.I., and Yosef, N. (2021). Multivi: Deep Generative Model for the Integration of Multi-Modal Data. Preprint at bioRxiv. https://doi.org/10.1101/2021.08.20.457057. |
| Cobolt | Gong, B., Zhou, Y., and Purdom, E. (2021). Cobolt: integrative analysis of multimodal single-cell sequencing data. Genome Biol. 22, 351–421. |
| scMM | Minoura, K., Abe, K., Nam, H., Nishikawa, H., and Shimamura, T. (2021). Scmm: Mixture-Of-Experts Multimodal Deep Generative Model for Single-Cell Multiomics Data Analysis. Preprint at bioRxiv. https://doi.org/ 10.1101/2021.02.18.431907. |
| Multigrate | Lotfollahi, M., Litinetskaya, A., and Theis, F.J. (2022). Multigrate: SingleCell Multi-Omic Data Integration. Preprint at bioRxiv. https://doi.org/10. 1101/2022.03.16.484643. |
| MOFA+ | Argelaguet, R., Arnol, D., Bredikhin, D., Deloro, Y., Velten, B., Marioni, J.C., and Stegle, O. (2020). Mofa+: a statistical framework for comprehensive integration of multi-modal single-cell data. Genome Biol. 21, 111–117. |
现存问题
- 需要在可扩展性、可解释性和灵活性进行权衡
- 完全数据驱动,不能充分利用生物学信息,例如基因注释和通路信息
功能
- 细胞聚类,识别细胞亚型
- 基于一个组学数据插补另一个组学数据
- 识别细胞类型特征和生物标志物
方法
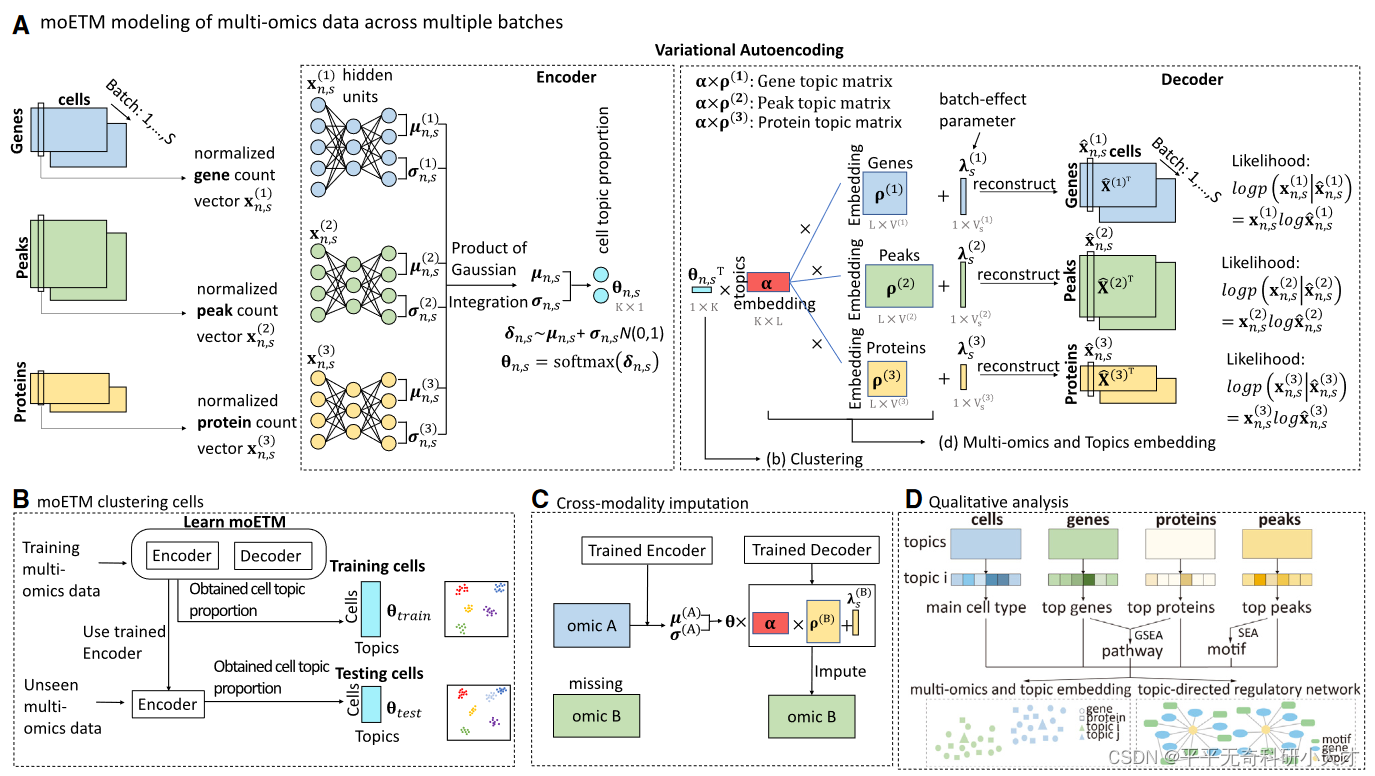
整体是VAE架构,但是他在Encoder和Decoer分别作了改进:
Encoder改进
- 假设每个组学数据分布符合K维独立的逻辑正态分布,这里采用K维高斯乘积(PoG)充分利用这些分布的信息,比之前的MoE得到更有效的变分推理。
- 前人方法对每个组学分别进行采样K维高斯变量然后平均化,这里只需从联合高斯采样一次,因此可以得到更鲁棒的结果。
- Topic解释:对联合高斯密度进行Softmax计算,生成的逻辑正态分布可视为细胞的主题混合。
Decoder改进
- 矩阵分解作为Decoder,把cell-by-feature matrices分解成shared cell-by-topic matrix,shared topic-embedding matrix和M(组学数量)个独立的feature-embedding matrices
- 引入组学特异性的批次移除因子λ,作为线性可加的批次特异性偏差
评价指标
生物保守性
- Adjusted Rand Index (ARI)
- Normalized Mutual Information (NMI)
批次效应移除
- k-nearest neighbor batch effect test (kBET)
- Graph connectivity (GC):衡量不同批次相同细胞类型之间的相似性,同时衡量生物保守型和批次移除效应
实验设置
随机分为训练集:测试集=6:4,重复500次
结果
多组学数据整合
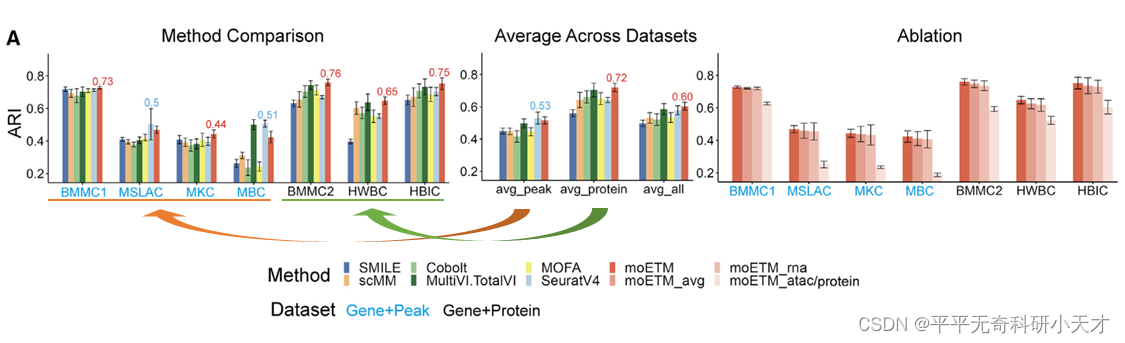
- 在4个peak-gene数据集上的平均指标第二,在3个gene-protein数据集上的平均指标第一,在所有7个数据集上平均指标第一
- moETM_* 为只利用组学*的数据进行训练和测试,与moETM结果对比表示,整合多组学数据比单组学数据得到更准确的结果
- moETM_avg 用分别从每个组学的高斯分布分别采样然后平均化代替PoG算法,结果降低了,说明PoG对于moETM起重要作用
cell-topic mixture可解释性
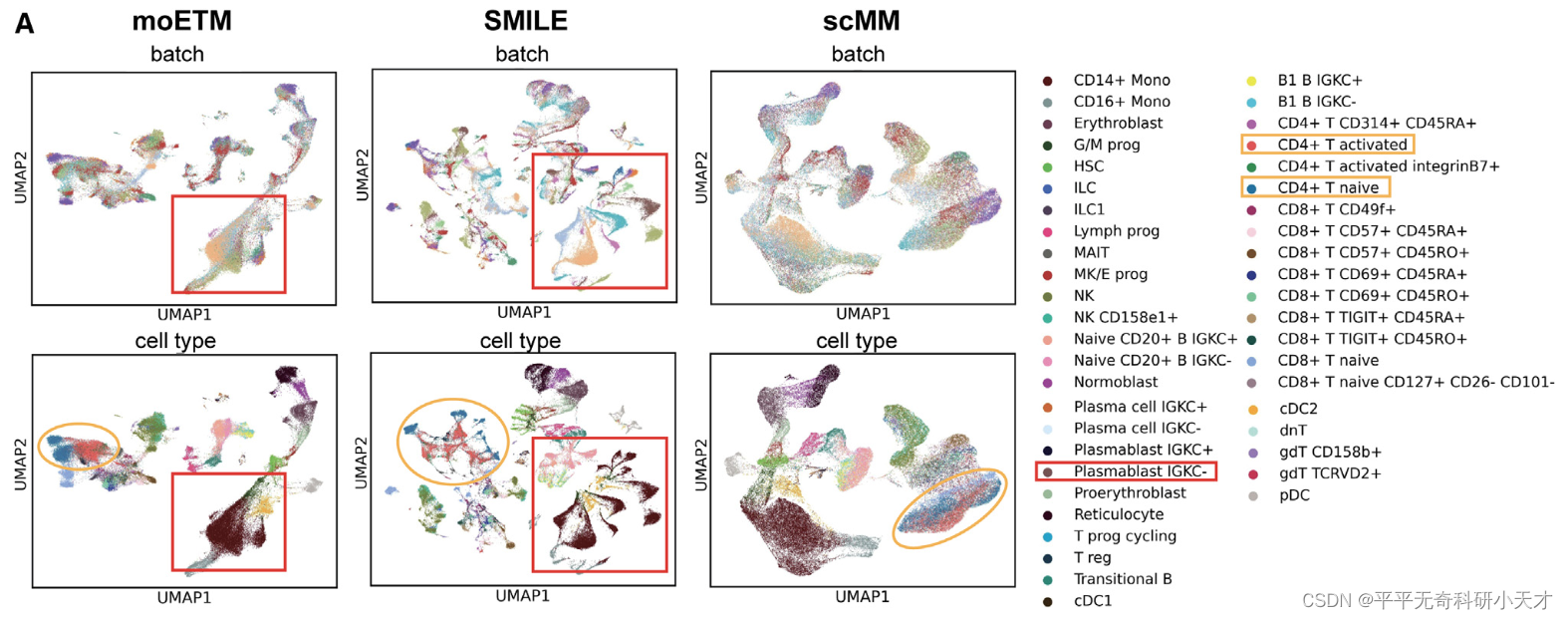
使用BMMC2数据集,把moETM训练得到的cell-by-topic matrix进行UMAP可视化,与其他方法得到的cell embdding可视化进行对比。
说明,cell-by-topic matrix既消除的BatchEffect,又识别了细胞类型。
组学翻译性能评估
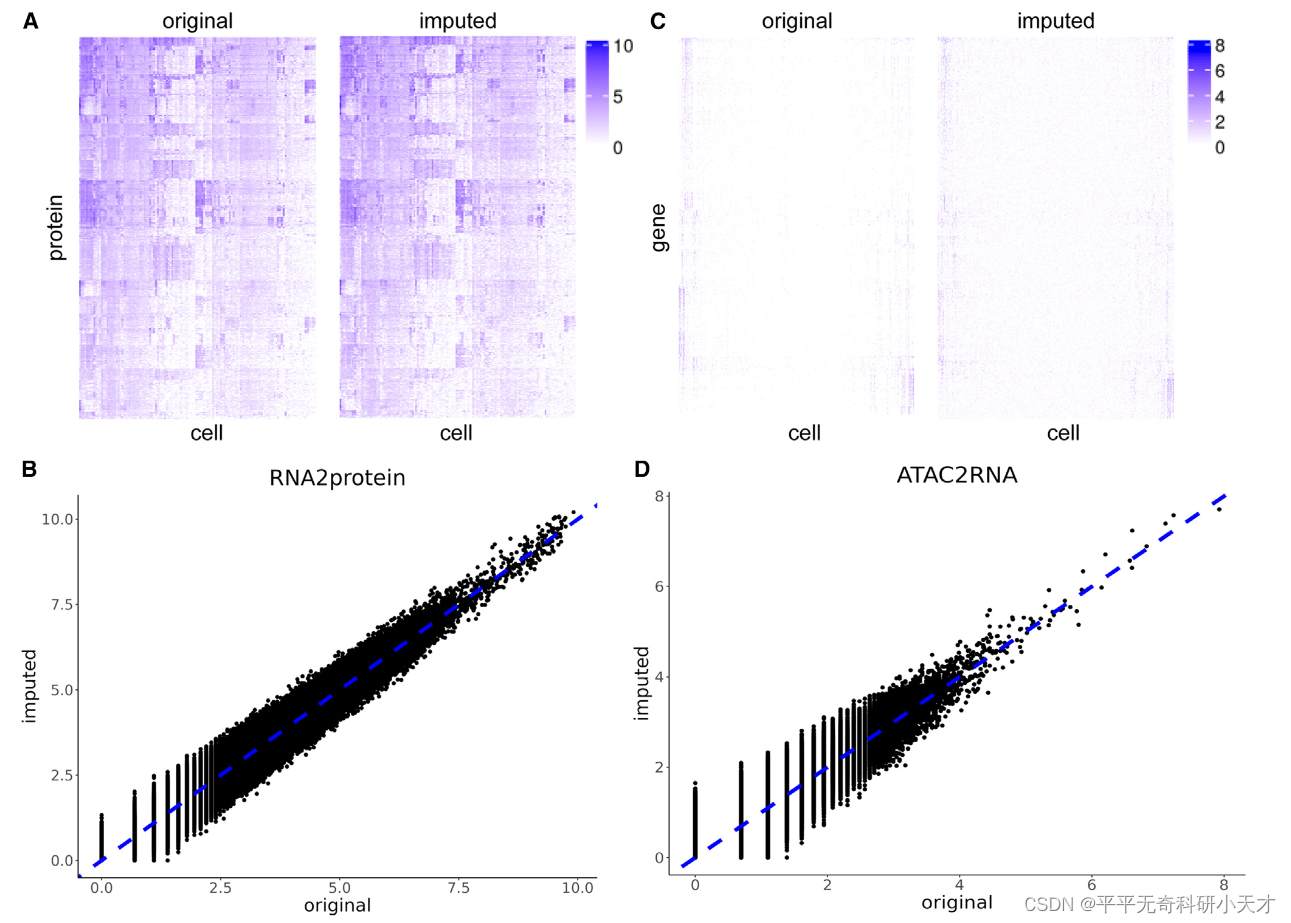
- 由A、B图看出,基于转录组数据翻译的蛋白数据与原始蛋白数据高度相似,且高度线性相关(PCC约0.95)
- 由C、D图看出,基于ATAC数据翻译的基因数据与原始基因数据高度相似,且高度线性相关(PCC约0.69)
- 由A、C图对比看出,相比蛋白表达数据,基因表达数据明显更稀疏。因此,基于ATAC翻译RNA比基于RNA翻译蛋白更难
- 实验结果的PCC(ATAC2RNA=0.69,RNA2ATAC=0.58,RNA2protein=0.95,protein2RNA=0.65),由此推断翻译任务难度RNA2protein < ATAC2RNA < protein2RNA < RNA2ATAC
RNA转录本、表面蛋白、染色质可及域调控关系研究
1. 验证同一主题下,top gene可以映射到top protein
过程
对于每个topic,计算134对基因和对应的翻译蛋白的 topic score 的Spearman correlation
结果
- 平均相关性在0.29
- 13个topic 相关性高于0.5
2. 跨主题验证gene-protein、peak-gene的调控关系
过程
如果一个peak在一个基因转录起始位点150k bp之内,则认为他们是匹配的。
查看匹配的peak-gene、gene-protein的相关系数分布
结果
查看匹配的peak-gene、gene-protein的相关系数分布显著高于0,并且和观测值得到的分布类似。
说明:该算法在整合的时候保留的调控相关性,且能反映原始数据特征
3. 验证重构的gene-protein、peak-gene更能反映组学相关性
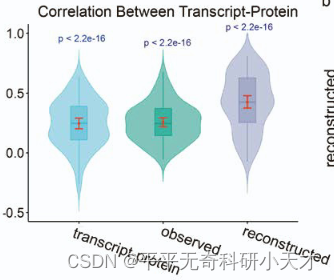
表明模型可以对观测数据的噪声进行降噪和混杂校正,更能反映单细胞中的不同组学的相关性
联合peak-gene分析细胞类型特异性通路和调控机制
分析细胞类型特异性通路
- 聚焦于一种类型的细胞(CD8+ T cells),将peak匹配到gene上,peak在一个基因转录起始位点150k bp之内,找到peak-neighboring genes。联合Topic score较高的的Top gene、Top peak联合分析
- 发现Top5 genes中3个与T细胞功能相关,Top5 peak对应的peak-neighboring genes中2个与T细胞功能相关,说明揭示了细胞类型特异性基因。
- 对Top5 genes、Top5 peak-neighboring genes进行通路富集(GSEA),得到的富集pathway与当前细胞类型相关,富集的基因集在当前细胞类型中显示出差异性表达(上调或下调),表明揭示了细胞类型特异性通路,及调控机制
分析细胞类型特异性motif
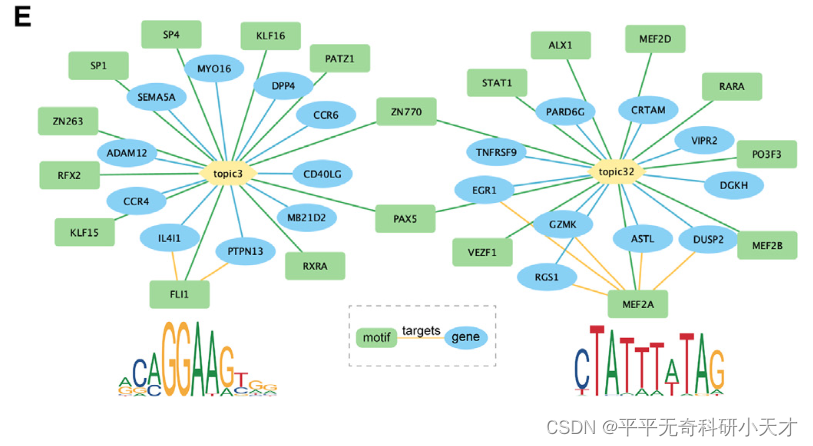
- 聚焦于一种类型的细胞(CD8+ T cells),从Ensembl database中,寻找根据Topic score排名的Top100 peaks对应的100个序列
- 将100个序列输入SEA算法,寻找这100个序列富集的motif
- 通过连接 Top genes,细胞类型、富集motif对应TFs 构造细胞类型特异性调控网络
- 通过ENCODE TF Targets dataset将已知的TF-genes用黄色线连接起来
- 结果表明:根据peak得到的motif在这种细胞类型中若干靶基因属于Top genes,说明模型识别了细胞类型特异性调控机制和motif特征
分析细胞类型特异性通路联合motif
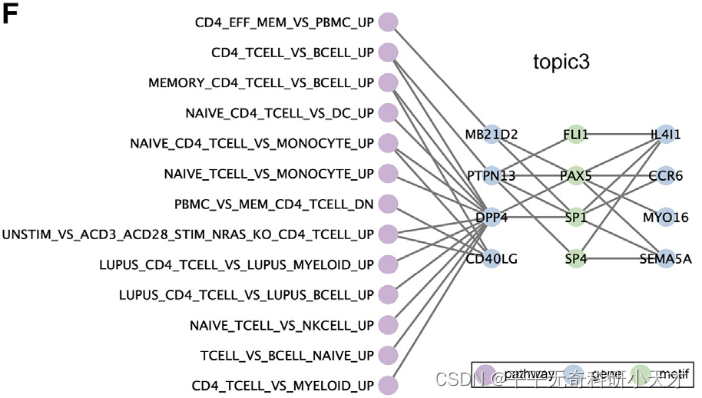
- 聚焦于一种类型的细胞(CD8+ T cells),通过连接 Top genes、富集motif对应TFs、富集pathway构建pathway-motif网络
- motif与Top genes根据ENCODE TF Targets dataset记录的调控关系连线
- 对比该类型的富集motif与相关的pathway(基因特异性表达,上调或下调),表示motif和pathway之间的调控关系一致性
补充
基因(蛋白)集富集过程
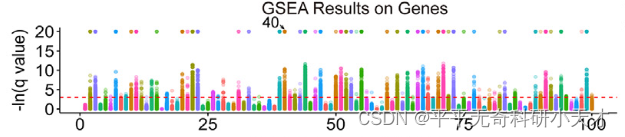
- 对于每个Topic,根据主题分数得到的rank gene list,通过运行GSEAPreranked函数从Molecular signatures database (MSigDB)查询2种基因集(免疫学特征基因集,基因本体生物过程)
- 对于过表达或低表达的基因计算富集分数(ES)
- 计算ES的统计学显著性
- 认为显著性 p-value<0.05 的基因集是显著的
- 图中每个颜色代表一个基因集(pathway),虚线以上的代表具有显著性。目的是说明每个Topic均可以显著性富集到基因集或pathway
基因与翻译蛋白表达负相关的可能原因
- 随机噪声可能会阻碍基因和蛋白质之间的相关性
- 单细胞水平的动态细胞过程(转录爆发、转录或翻译延迟)可引起细胞之间的差异,导致相关性降低
- 其他生物过程的影响压倒了转录的影响(转录后翻译的影响超过了蛋白质合成)
- mRNA降解速度超过蛋白质合成速度
寻找细胞标志物的资源–CellMarker数据库
Zhang, X., Lan, Y., Xu, J., Quan, F., Zhao, E., Deng, C., Luo, T., Xu, L., Liao, G., Yan, M., et al. (2019). Cellmarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 47, D721–D728.
寻找TF对应靶基因的资源–ENCODE Transcription Factor Targets
The ENCODE Project Consortium (2011). A user’s guide to the encyclopedia of dna elements (encode). PLoS Biol. 9, e1001046.
ENCODE Project Consortium; and Pachter, L. (2004). The encode (encyclopedia of dna elements) project. Science 306, 636–640.