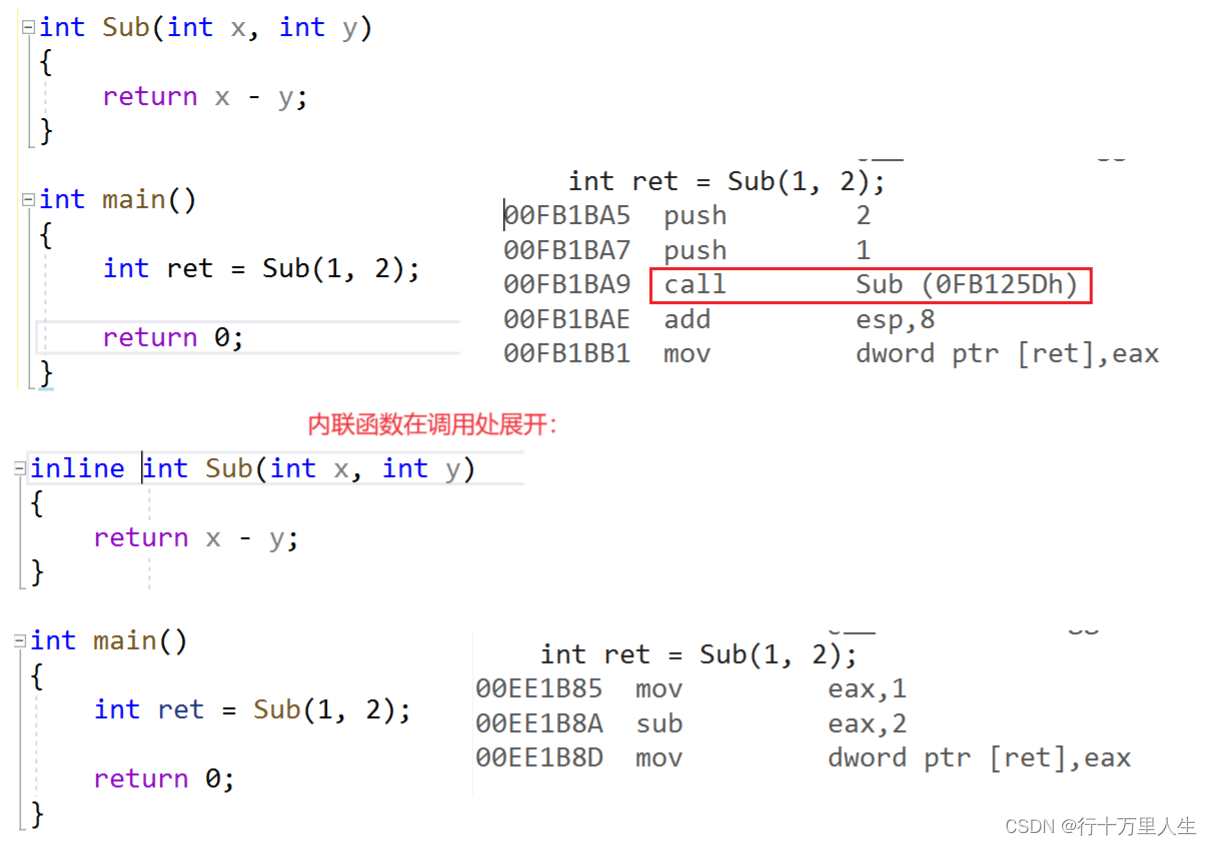
文章目录
- RNA转录重建
- 1 先简单介绍一下测序相关技术
- 2 Map to Genome Methods
- 2.1 Step1 Mapping reads to the genome
- 2.2 Step2 Deal with spliced reads
- 2.3 Step 3 Resolve individual transcripts and their expression levels
- 3 Align-de-novo approaches
- 3.1 Step 1: Generate all k-mers and link them together
- 3.2 Step 2: Collapse the De Bruijn Graph
- 3.3 Step 3: Resolve graph, quantify isoform abundance
- 4 Differential expression analysis
来自Manolis Kellis教授(MIT计算生物学主任)的课
油管链接:6.047/6.878 Lecture 7 - RNA folding, RNA world, RNA structures (Fall 2020)
预习,一定要先看这个:【生信小课堂】如何直观的理解RNA-seq分析流程
本节课分为三个部分,本篇笔记是第二部分RNA structures。
本部分深入研究了RNA转录的重构和测序技术。从RNA转录重构的基础开始,探讨了现代测序技术的发展和应用。此外,还讲述了基因组映射技术如何帮助研究者更好地理解RNA转录,以及de-novo对齐和差异表达分析的重要性
RNA转录重建

1 先简单介绍一下测序相关技术
- 用于基因表达分析的两种主要技术:Microarray技术 和 RNA-Seq技术
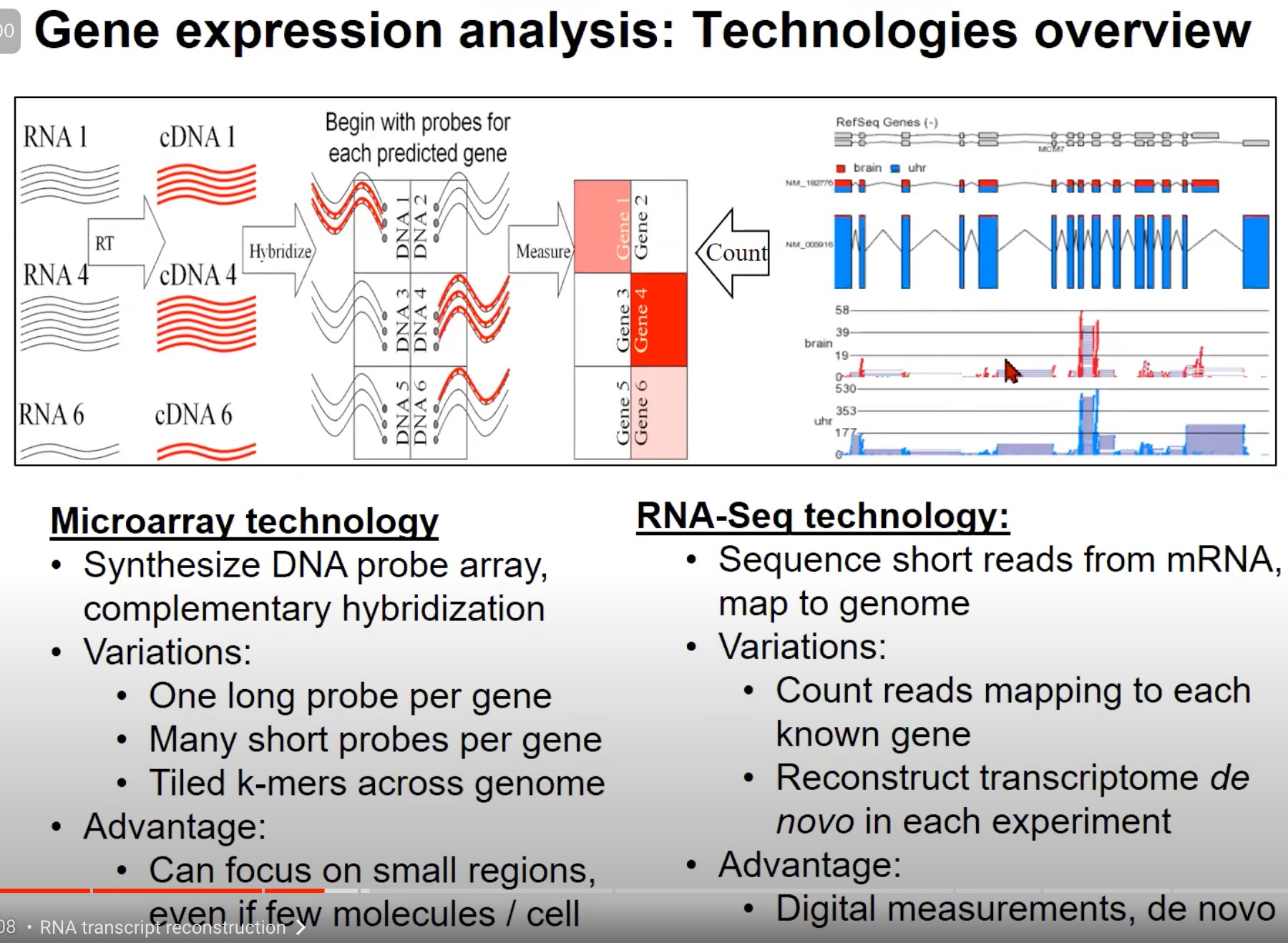
1. Microarray技术 (芯片技术):
- 原理:Microarray技术使用DNA探针阵列进行互补杂交。对于每个预测的基因,你首先用一个特定的探针去检测它。
- 变化:
- 每个基因一个长探针
- 每个基因多个短探针
- 整个基因组的倾斜的k-mers(k个核苷酸的片段)
- 优势:即使在每个细胞中只有少量的分子,也可以关注小的区域。
2. RNA-Seq技术:
- 原理:RNA-Seq技术通过测序从mRNA得到的短读数并将它们映射到基因组。
- 变化:
- 计数映射到每个已知基因的读数
- 在每次实验中都重新构建转录组 (de novo)
- 优势:这是一个数字化的测量方法,可以从头开始(de novo)进行。
两种技术都可以提供关于基因表达的信息,但它们的方法和优势各不相同。Microarray是一种更传统的技术,它依赖于预先知道的基因信息,而RNA-Seq提供了一种更全面、数字化的方法来测量和分析基因表达。
接下来介绍一下RNA-Seq实验数据生成的流程。
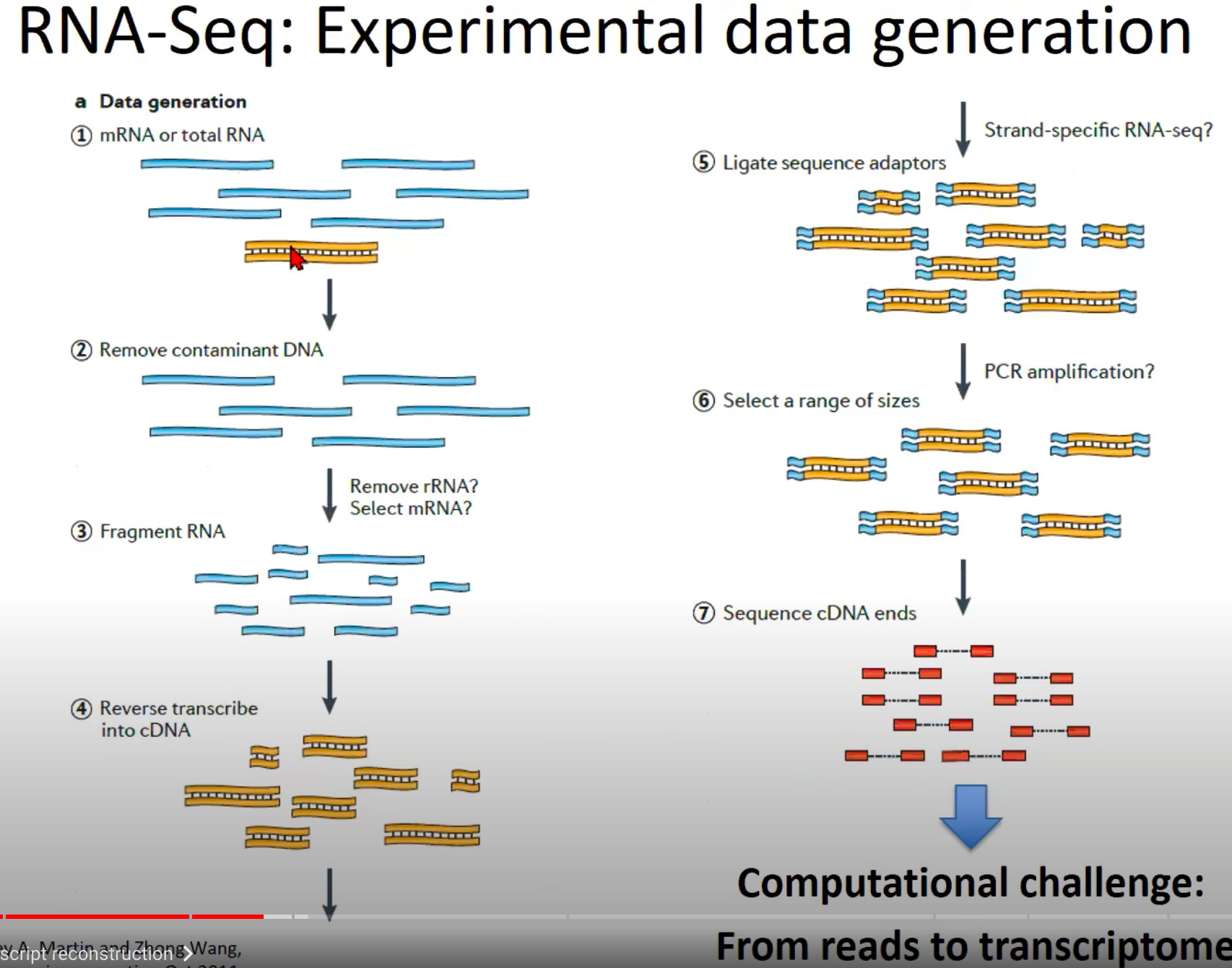
这个图展示了RNA-Seq实验数据生成的流程。下面是该流程的逐步解释:
- mRNA或总RNA提取:首先,从细胞或组织样本中提取mRNA或总RNA。mRNA是经过剪切的、成熟的RNA,而总RNA包括mRNA和其他类型的RNA(如核糖体RNA、转运RNA等)。
- 去除污染的DNA:由于RNA提取过程可能会带有DNA污染,因此需要将这些DNA去除,确保只对RNA进行测序。
- RNA片段化:为了使RNA适合于测序,RNA被打断成更小的片段。
- 反转录成cDNA:RNA片段通过反转录酶被转化为互补DNA(cDNA)。cDNA是双链的,与原始的RNA片段对应。
- 连接测序接头:为了使cDNA片段能在测序机上被读取,需要将特定的接头序列连接到cDNA的两端。
- 选择一定范围大小的片段:通过大小选择步骤,选择特定长度范围内的cDNA片段进行下一步测序。
- 测序cDNA末端:最后,选择的cDNA片段在测序机上被测序。这通常涉及到只测序cDNA片段的一端或两端(称为单端测序和双端测序)。
- Challenge:从读数到转录组的重建。这意味着,一旦获得了测序读数,我们面临的主要挑战是如何将这些短的测序读数组装回原始的RNA序列,并从中识别和量化基因和其他转录单位。
Tips:
跟Illumina测序平台的特点有关。以下是为什么现在的测序主要关注小片段并且通常只在片段的开头和末端测序的原因:
- 测序深度与覆盖率:通过测序小片段的DNA,可以获得较高的测序深度和更均匀的基因组覆盖率。这对于确保数据的准确性和鉴定低频率的变异非常重要。
- 开头和末端的测序:对于某些应用,例如配对末端测序,只需要测序DNA片段的两端。这使得可以间接获得长片段的信息,如插入、缺失和结构变异。
- 准确性和质量:测序小片段可以提高测序准确性,因为测序错误的累积可能会在长片段的后续部分增加。
- 从RNA-Seq读取中重构转录本的两种方法
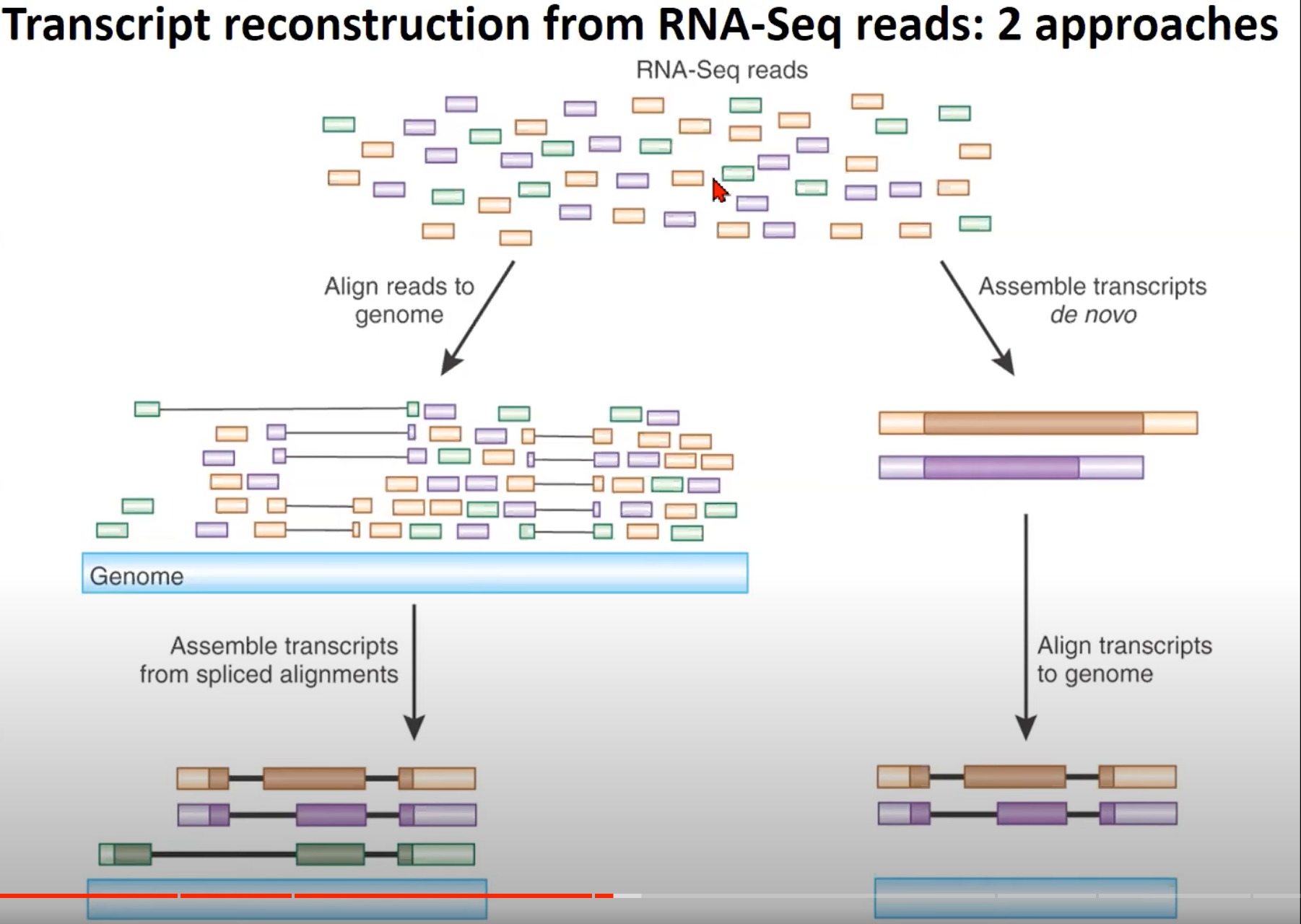
- RNA-Seq读取:RNA-Seq产生的多种读取。这些读取是从RNA样本中获得的,并代表了不同的转录本片段。
- 对齐读取到基因组:RNA-Seq读取首先被对齐到参考基因组。这是重构转录本的第一步。这样做的目的是确定每个读取在基因组中的位置。
- 从拼接对齐中组装转录本:经过对齐后,根据拼接对齐的结果,读取会被组装成完整的转录本。这些转录本表示了原始RNA样本中存在的不同的RNA分子。
- de novo组装转录本:另一种方法是直接从RNA-Seq读取中de novo(从头开始)组装转录本,而不是首先对其进行对齐。这种方法特别适用于没有可用的参考基因组的物种。
- 将转录本对齐到基因组:完成de novo组装后,为了进一步验证和注释,这些转录本可以与参考基因组进行对齐。
2 Map to Genome Methods
“Tuxedo Suite”工具,将RNA-seq对齐到参考基因组
当从生物样本中获取DNA或RNA测序数据时,您会得到数百万或数十亿的短测序读取。为了理解这些读取的含义,必须知道它们在基因组上的位置。这样,可以确定读取来自基因组的哪个部分,例如特定的基因或非编码区域

“Tuxedo Suite”是一个流行的RNA-seq数据分析pipeline。由一系列软件组成,这些软件按照特定的顺序串联起来,以完成从原始测序数据到基因和转录本表达量估计的整个分析过程。
- Bowtie:是一个超快的、内存效率高的短读取对齐工具,用于将测序读取对齐到大型基因组。
- TopHat:使用Bowtie为基础的对齐引擎,但经过优化,可以发现由于外显子剪接导致的读取分割。因此,TopHat可以对齐经过剪接的RNA-Seq读取。
- Cufflinks:用于组装由TopHat对齐的读取,并通过统计模型估计基因和转录本的表达量。
- Cuffdiff:是Cufflinks的一个部分,用于在多个样本之间比较基因和转录本的表达量,以确定表达差异。
- CummeRbund:用于对Cufflinks和Cuffdiff的输出进行后处理和可视化。
接下来是详细介绍
2.1 Step1 Mapping reads to the genome
transcript reconstruction:Map-to-Genome methods 映射到基因组
转录本重建是一个过程,目标是根据RNA-Seq数据确定转录起始和终止的精确位置,以及由哪些外显子构成。简而言之,它的目的是定义一个给定基因的所有可能的转录本,并确定哪些区域在RNA分子中是连续的。
RNA-Seq技术产生了数以百万计的短序列读取,这些读取来自样本中表达的RNA分子。转录本重建的挑战在于,从这些分散的短读取中,正确地组装或“重建”出原始的RNA分子(即转录本)。
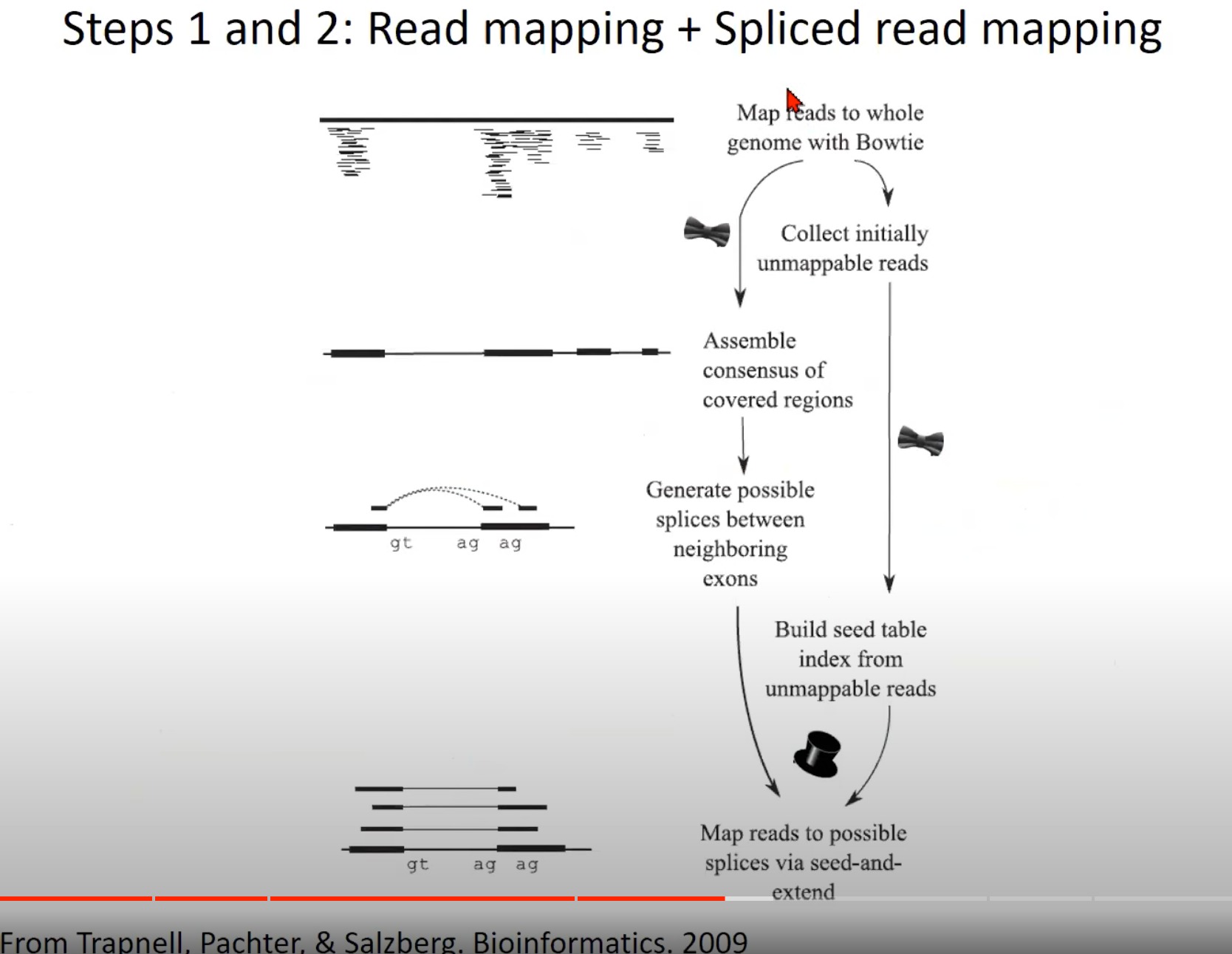
RNA-Seq数据分析的前两步:读取映射和剪接读取映射。
-
Map reads to whole genome with Bowtie: 使用Bowtie工具将RNA-Seq读取直接对齐到整个基因组。这是RNA-Seq数据分析的初步,目的是找到每个读取在基因组上的位置。
-
读取映射
-
Assemble consensus of covered regions: 基于已经映射到基因组的读取,这一步试图组装覆盖区域的一致序列。
-
Generate possible splices between neighboring exons: 根据前面的结果,这一步尝试预测可能的剪接位点,即RNA在哪里被切割和再连接,从而形成成熟的mRNA。
- 这步在Build seed table index from unmappable reads之前
-
-
剪接读取映射
-
Collect initially unmappable reads: 在第一次对齐尝试中,有些读取可能不能直接对齐到基因组,这些被称为“初步不可映射的读取”。
- 原因可能是这些读取跨越两个或多个外显子,因此它们包含了RNA的剪接位点。(在基因组上的位置是断开的,两个外显子之间有一个内含子)
-
Build seed table index from unmappable reads: 使用初步不可映射的读取建立一个种子表索引。这是为了帮助在下一步中找到这些读取可能对齐的正确位置。
-
-
Map reads to possible splices via seed-and-extend: 使用种子和扩展策略对初步不可映射的读取进行对齐。这意味着首先找到一个小的匹配区域(种子),然后尝试扩展这个匹配,直到找到完整的对齐或达到一个预定的限制。
图中的“gt ag ag”可能表示典型的剪接位点,在真核生物中,剪接通常发生在“gt…ag”序列上。内含子的起始部分是“GT”,而结束部分是“AG”。
补充内容:
【生信常识】二代测序的比对算法浅析
B站原理推导
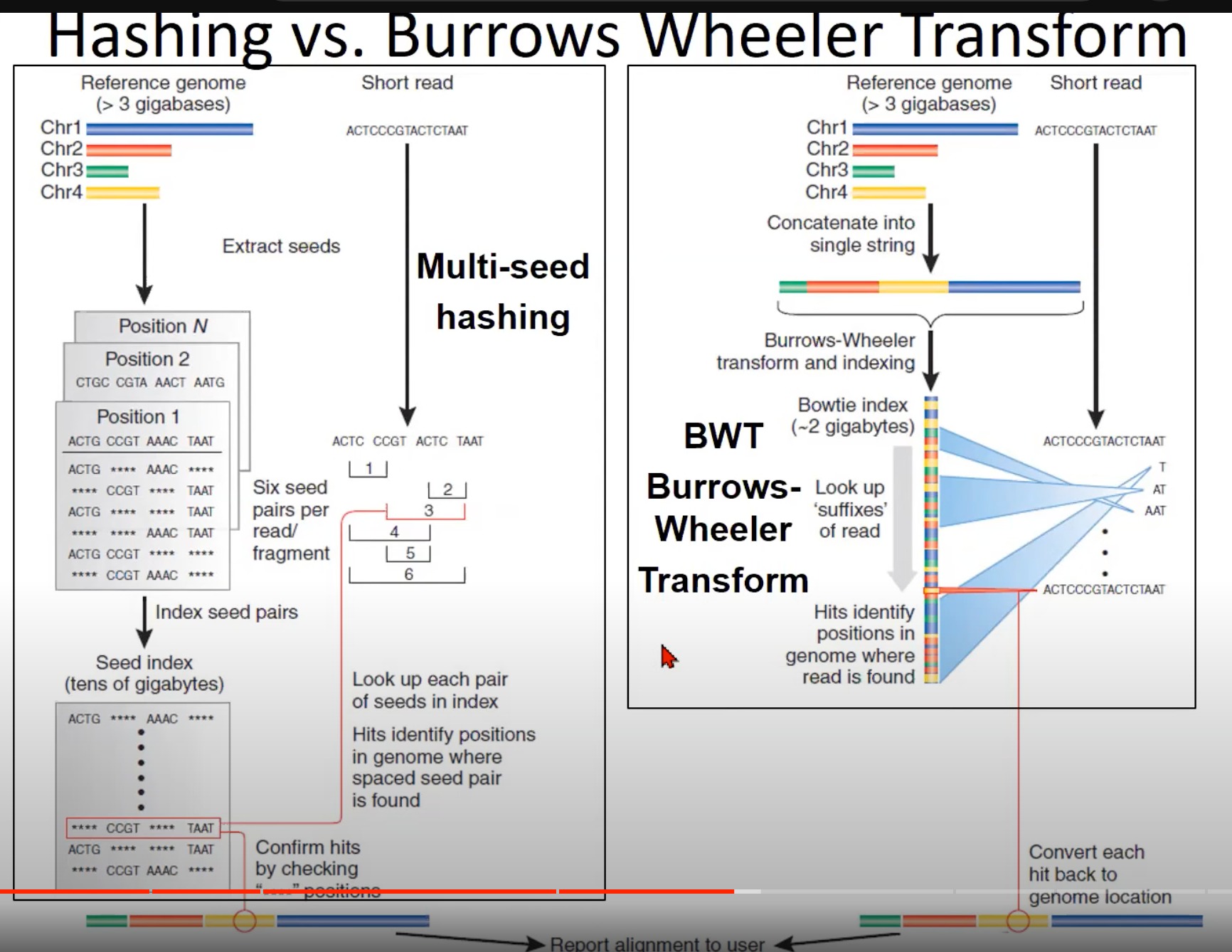
两种不同的短读取对齐方法:哈希法(Hashing)和Burrows-Wheeler Transform (BWT)。
- 哈希法 (左侧):
- 从参考基因组中提取“种子”(较短的连续DNA片段)。
- 每个种子对应于参考基因组上的一个位置。
- 这些种子被索引,即将每个种子和其在参考基因组上的位置相关联。
- 当一个短读取被给出时,它也被分解为种子。然后查找这些种子在索引中的位置,从而确定短读取在参考基因组上的位置。
- 这种方法的挑战在于需要大量的存储空间来存储种子索引,尤其是当处理较大的基因组时。
- Burrows-Wheeler Transform (BWT) (右侧):
- 参考基因组首先被转换为一个字符串。
- 使用Burrows-Wheeler变换和后续的索引,将这个字符串转换为BWT索引。BWT是一个有效的算法,使得查找短读取在参考基因组中的位置变得更快、更节省空间。
- 对于给定的短读取,使用BWT查找其在参考基因组中的位置。
- 与哈希法相比,BWT方法使用的存储空间更少,速度也更快。
- Bowtie和Bowtie 2是使用BWT的流行的短读取对齐工具。
geneXplain 平台中的 RNA-seq 预处理:读取对齐
- Bowtie2和动态规划比对算法的关系
- Bowtie2主要是用于高通量数据测序比对,将reads贴到参考基因组上的一个range中。
- 定位好后
- 就可以在这个range内可以再和reads使用Smith-Waterman算法/Needleman-Wunsch算法进行精确比对,打分。
- 打分有什么用呢
- 我推测是如果一个read可以map到参考基因组的多个位置,就可以对其进行打分对比,选取最有可能map到的位置
具体原理我也不是很懂,有待进一步学习
- 我推测是如果一个read可以map到参考基因组的多个位置,就可以对其进行打分对比,选取最有可能map到的位置
2.2 Step2 Deal with spliced reads
处理剪接片段
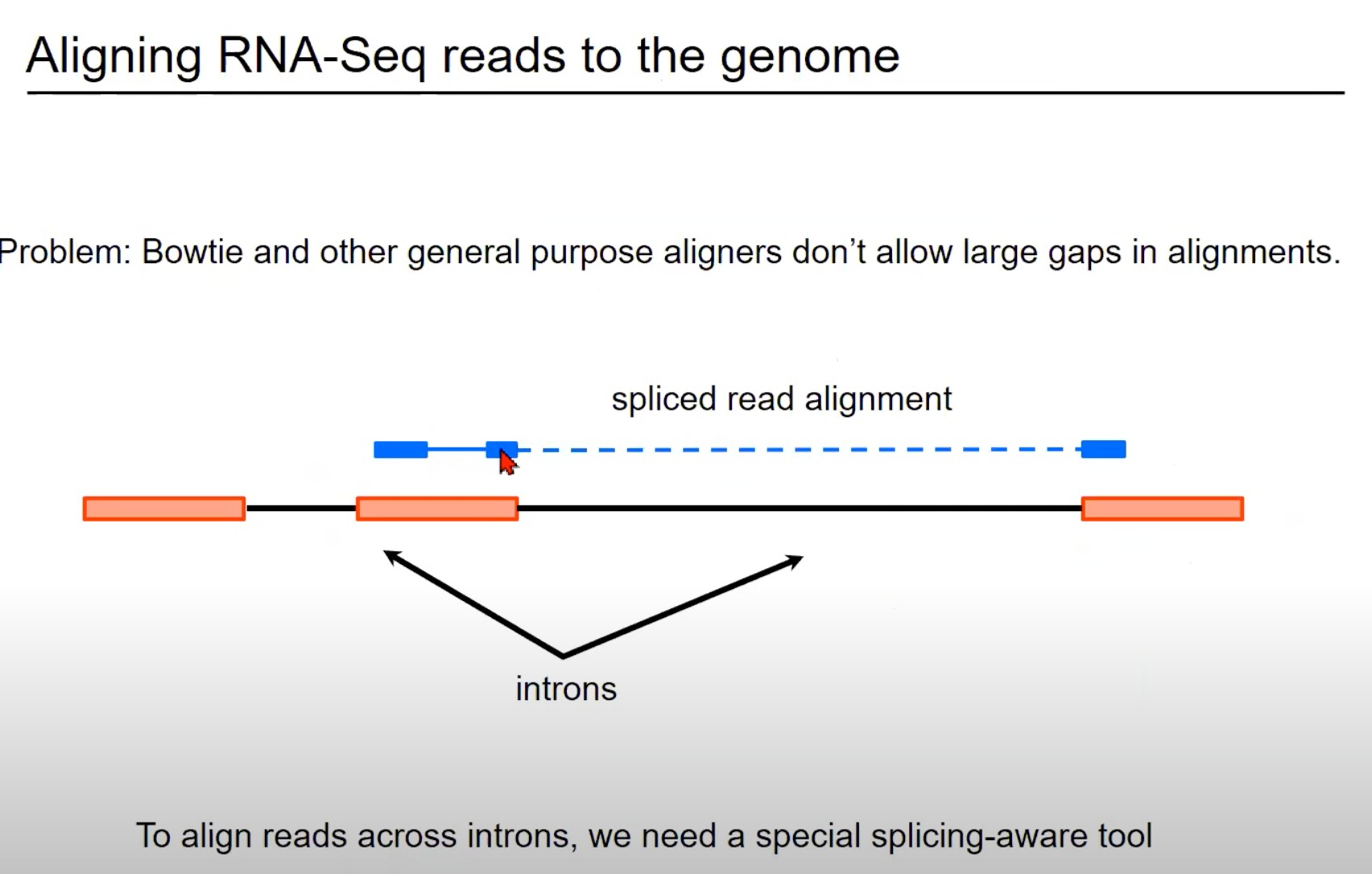
蓝色部分:这表示从RNA-Seq实验中获得的读取。
在这个例子中,这个读取跨越了一个内含子(intron),因此被称为剪接读取(spliced read)。您可以看到读取的一部分与一个外显子(exon)对齐,而读取的另一部分与另一个外显子对齐。
橙色/红色部分:外显子
标准的对齐工具,如Bowtie,通常不允许对齐中有大的间隙。但是,当处理RNA-Seq数据时,这样的间隙是常见的,因为读取可能会跨越一个或多个内含子。因此,需要一个能够识别和正确处理这种剪接事件的特殊对齐工具。
TopHat对齐RNA-Seq读取
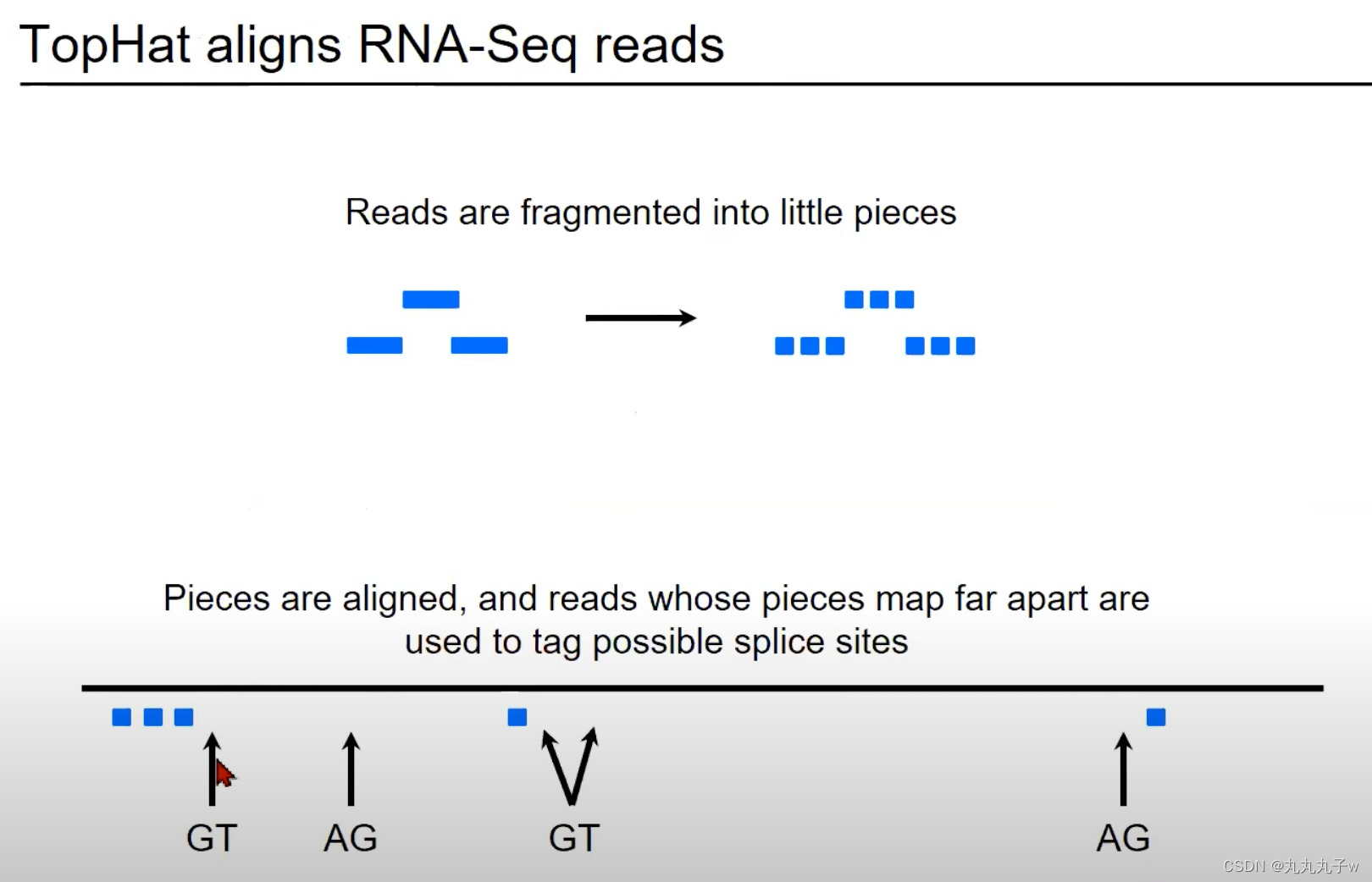
TopHat是一个专门为RNA-Seq数据设计的对齐工具,它能够检测并处理剪接事件。
- 读取的碎片化:
- 图中的蓝色矩形代表从RNA-Seq实验中获得的读取。这些读取首先被分割成更小的片段。
- 对齐片段并标记可能的剪接位点:
- 一旦碎片化完成,这些小片段就会被对齐到参考基因组。
- 如果某些片段在基因组上相对远离对齐,那么它们之间的区域可能是一个剪接位点。
- 在图中,两个矩形之间的箭头指向了"GT"和"AG",这些是真核生物常见的剪接信号。
- 这些片段可以提供有关可能的剪切位点的额外信息。
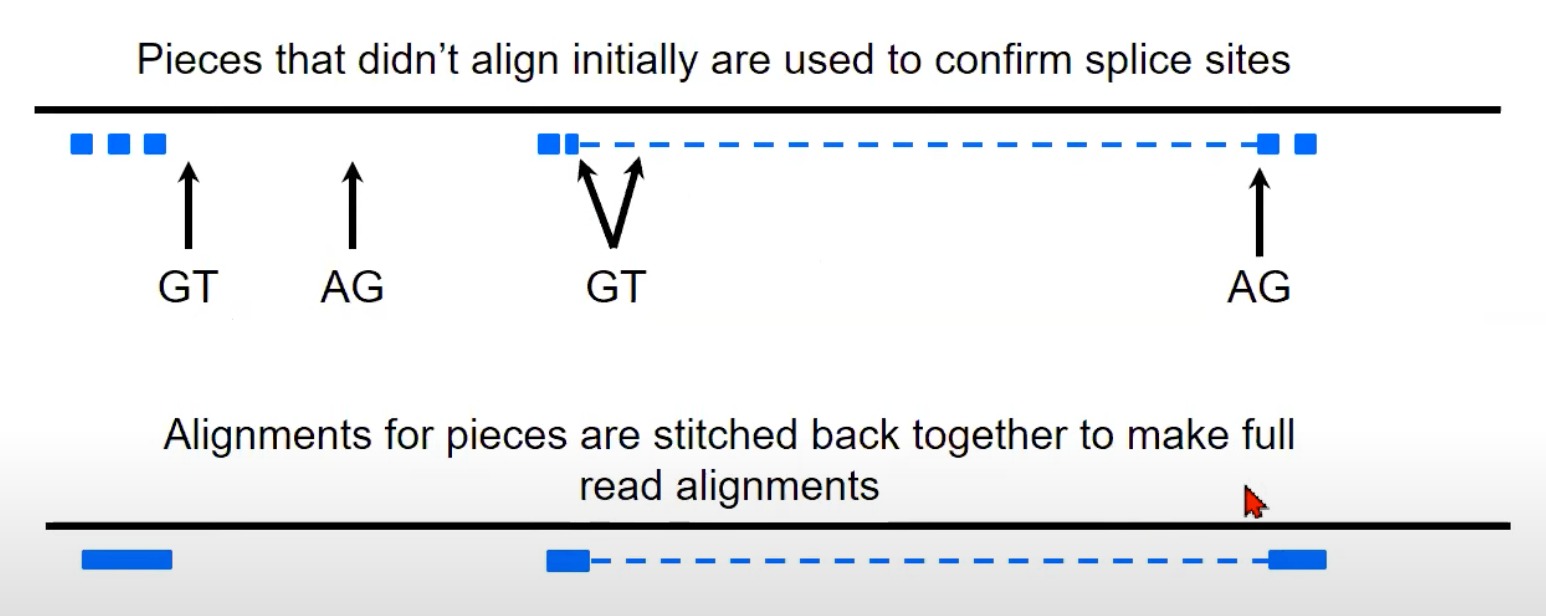
- 重组完整读取: 在确认了剪切位点之后,各个对齐的片段(即使它们在基因组上是分开的)会被“缝合”在一起,从而重建出原始的RNA读取。
- 图中显示的虚线表示原始的RNA读取可能会被分割成小片段,但经过处理后,可以将这些片段重新组合成完整的读取。
- 多看看这个示意图就懂了
TopHat使用这种策略来确定读取可能跨越的内含子位置,并进行适当的剪接对齐。
2.3 Step 3 Resolve individual transcripts and their expression levels
- 在Step1中,我们将测序读取映射到基因组上。
- 在Step2中,我们处理了跨越外显子剪切位点的读取。
- 第二步的意义是确定了很多跨越内含子的读取的映射问题
- 到了Step3,我们需要确定每个独立的转录本及其表达水平。
- 基因表达水平和测序读取数量之间的关系
- 为什么需要纠正基因的总长度,考虑基因的长度来计算表达水平
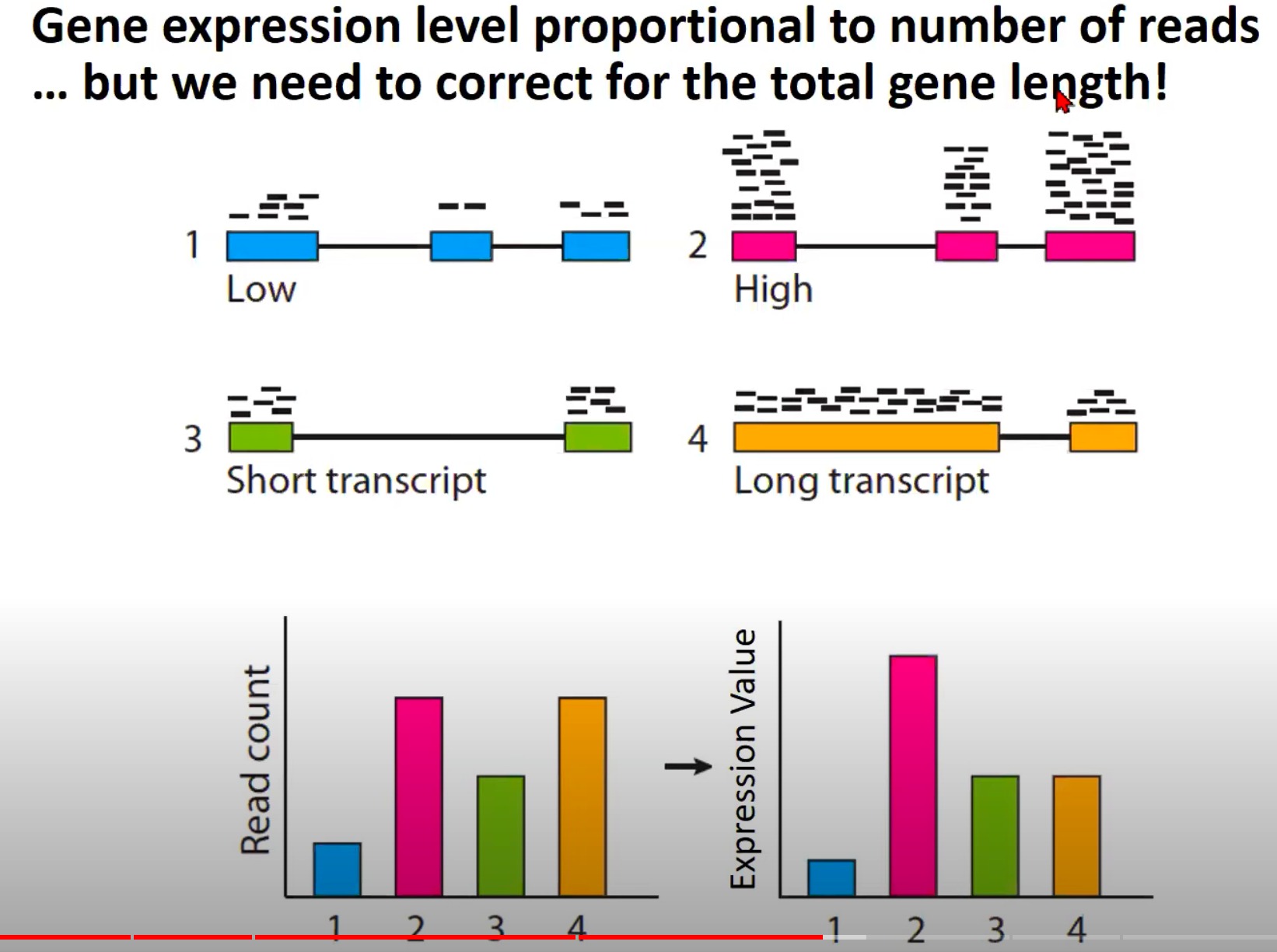
- Low (低表达): 这是一个低表达的基因,所以只有少量的读取与其对齐。
- High (高表达): 这是一个高表达的基因,有很多的读取与其对齐。
- Short transcript (短转录本): 这是一个短的基因转录本,尽管它可能有相对较高的表达,但由于其短的长度,只有少量的读取与其对齐。
- Long transcript (长转录本): 这是一个长的基因转录本,即使****,也可能有大量的读取与其对齐,因为它的长度更长。
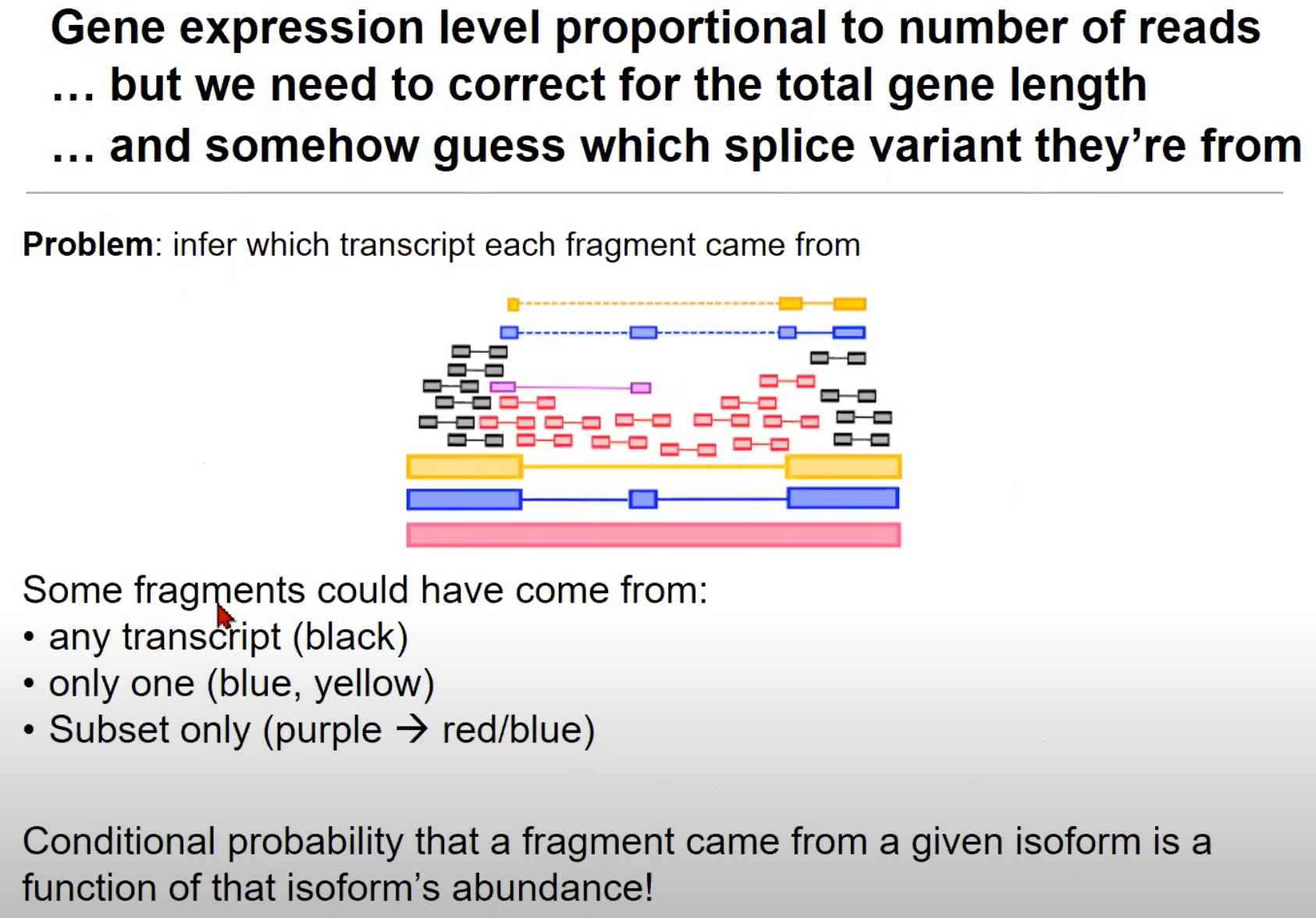
- 猜测每个片段来源于哪个拼接变体(Splice Variants):来自一个基因的读取可能来源于多种拼接变体。
- 拼接变体就是转录本
- 一个基因通过RNA拼接过程生成的不同mRNA分子。一个基因可以通过不同的方式拼接其RNA,从而产生不同的mRNA分子,这些mRNA分子可以翻译成不同的蛋白质。
- 红色的转录本可能包含了某些外显子,而这些外显子在蓝色和黄色的转录本中被省略了。
- 片段可能来自的转录本:
- 任何转录本:黑色的片段可以来自任何转录本。
- 只来自一个:蓝色和黄色的片段只能来自一个特定的转录本。
- 黄对黄、蓝对蓝是因为RNA-seq读取出来的就是那样,黄色是没有中间那一块的
- 只来自子集:例如,紫色的片段可能只来自红色或蓝色的转录本。
- 片段来自某一同构体的条件概率:这意味着我们估计一个片段来自特定同构体(或拼接变体)的概率是与那个同构体的丰度相关的。例如,如果一个同构体的丰度很高,那么来自它的片段的概率也会很高。
- 意思是这个转录本/同构体,在总的RNA中占比高,就是基因大概率都转录成这个样子的
- 转录本定量
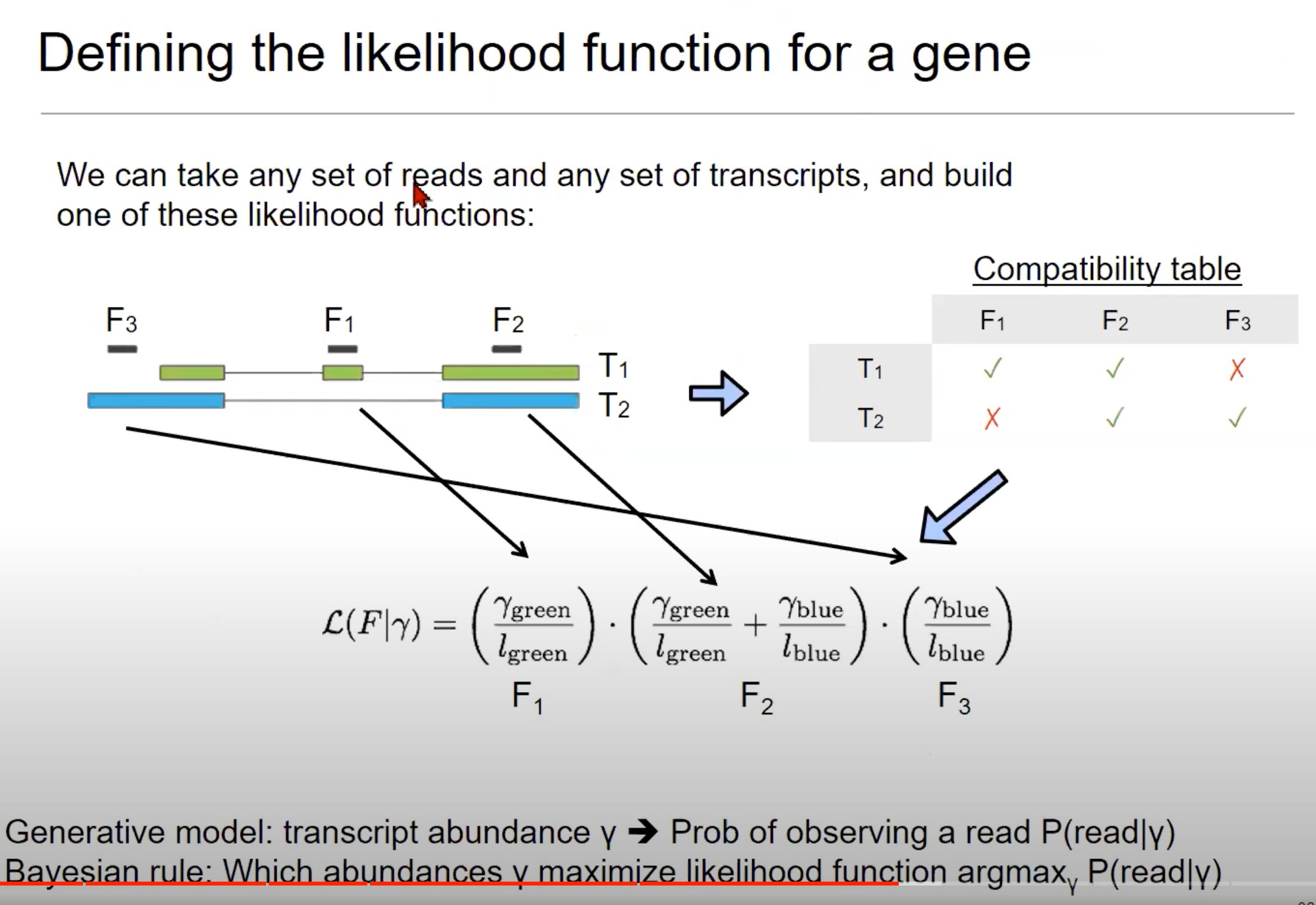
-
为一个基因定义似然函数。
- 似然函数是统计学中的一个概念,用于描述在给定参数下观察到数据的可能性。
- 我们要确定给定的RNA测序读取(reads)最有可能来自哪些转录本(transcripts)。
- 这是RNA-Seq数据分析中的一个关键步骤
-
这里的目标是确定每个转录本的丰度,即每个转录本在总RNA中的比例。
- 我们要从结果去倒推原因,通过观察到的短reads去推测丰度,使用贝叶斯法则
- 我们有一个关于转录本丰度的“先验”概念,然后使用观测到的读取数据来更新这一先验概念,得到转录本丰度的“后验”估计。
- 我们要从结果去倒推原因,通过观察到的短reads去推测丰度,使用贝叶斯法则
-
逐步解析
- 读取和转录本:图中展示了三个读取(F1, F2, F3)和两个转录本(T1, T2)。这些读取是RNA-Seq实验中从转录本中获得的小片段。
- 兼容性表格(Compatibility table):这个表格显示了哪些读取可以与哪些转录本相匹配。例如,F1可以匹配T1但不能匹配T2,而F3可以匹配T2但不能匹配T1。
- 每个读取(例如F1)对应的概率是考虑它来自各个转录本的可能性。例如,如果F1与T1和T2都兼容,则其对应的概率会是与T1的丰度有关的项和与T2的丰度有关的项的和。
- 似然函数:似然函数是基于读取与转录本的匹配程度来定义的。
- 公式中的γ代表转录本的丰度(abundance),即每个转录本在整体表达中的比例。
- l l l代表读取的长度。
- 这个函数试图解释读取来自每个转录本的可能性。
- 公式中的分数形式表示条件概率,例如给定绿色部分的丰度,观察到F1的概率。
- 通过将每个读取的概率相乘,我们可以得到所有读取的联合概率,从而优化转录本的估计丰度。
- 生成模型和贝叶斯规则:
- 生成模型:描述了给定转录本丰度y时,观察到一个读取的概率。这是根据上述似然函数定义的。
- 贝叶斯规则:这是一个用于估计最可能的转录本丰度的方法。这里使用的是
argmax函数,意思是找到一个丰度γ,使得观察到的读取的概率P(read|γ)最大。
-
目前我也处于一个似懂非懂的状态,
期待补充,反正就是利用现有的观测数据short-reads,去倒推一个转录本丰度,也就是这个转录本在总RNA中的占比
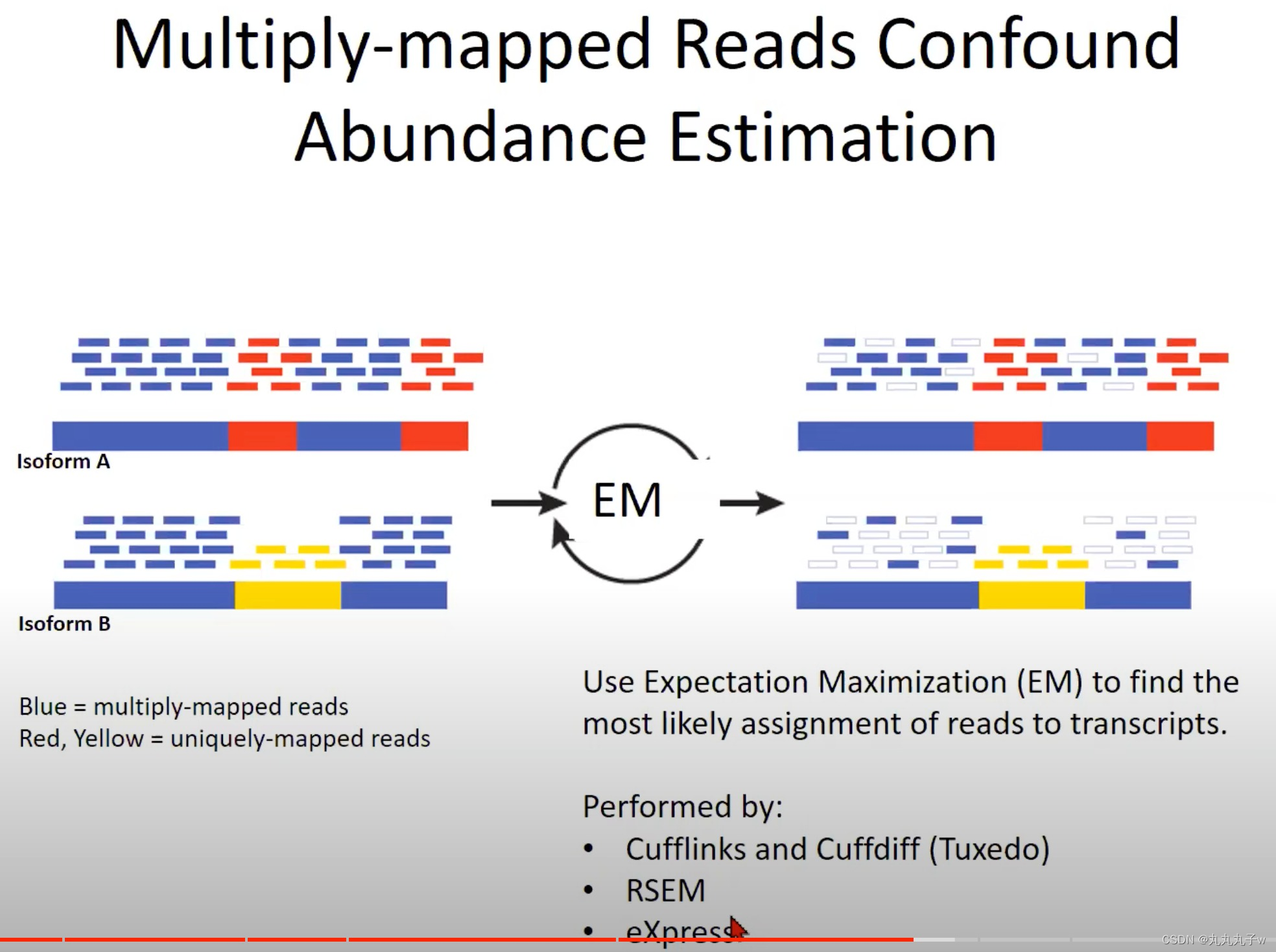
现实的情况更加复杂,多次映射的读取(Multiply-mapped Reads)会混淆丰度估计
- 多次映射的读取与唯一映射的读取:
- 多次映射:一个读取可能与基因组中的多个位置都有很高的相似度
- 因为基因组中存在很多重复的序列,或者有多个非常相似的基因或转录本。
- 蓝色表示的是多次映射的读取。这意味着这些短的测序片段可以映射到基因组上的多个位置。由于这个原因,它们不能确定地被分配到一个特定的转录本。
- 红色和黄色表示唯一映射的读取。这些读取在基因组上有一个唯一的位置,所以我们可以确定地知道它们来自哪个转录本。
- 多次映射:一个读取可能与基因组中的多个位置都有很高的相似度
- 期望最大化 (Expectation Maximization, EM):
- 为了解决多次映射的读取问题,我们使用了一个统计方法叫做期望最大化 (EM)。这个方法试图找到最有可能的方式将读取分配到转录本上。
- “Isoform A”和“Isoform B”以及它们对应的多次映射和唯一映射的读取。
- Isoform:转录本
- 经过EM处理后,读取被重新分配(如图的右侧所示),以便我们能得到对每个转录本的更准确的丰度估计。
EM算法原理,具体请b站
- 初始化:
- 首先,需要为每个转录本分配一个初步的丰度估计。这通常是随机的,或者基于一些其他的先验知识。
- 期望步骤 (E-step):
- 在这一步,我们使用当前的转录本丰度估计来计算每个多次映射的读取被分配到每个可能的转录本上的概率。
- 最大化步骤 (M-step):
- 基于上一步计算的概率,我们更新转录本的丰度估计。这是通过重新分配多次映射的读取来完成的,使得转录本的丰度估计与观察到的数据最为一致。
- 迭代:
- 重复E-step和M-step,直到转录本的丰度估计收敛(即变化非常小或不变)。
- 说实话,这部分实在难懂,就跟K-means聚类对比了一下
- 似然函数:在EM算法中,似然函数描述了给定当前参数估计(例如转录本的丰度)时,观察到的数据(读取)的概率。这与K均值聚类中每个点到其所属聚类中心的距离类似。在K均值中,我们希望最小化所有点到其聚类中心的距离之和。而在EM中,我们希望最大化似然函数。
- 丰度:转录本的丰度估计与K均值中的聚类中心类似。丰度描述了一个特定转录本存在的“强度”或“量”,而聚类中心是代表其所属群组的数据点的平均位置。
- 短读取:短读取在这里可以被视为数据中的“点”。
- 转录本:转录本在这里可以被视为“聚类”。
K均值聚类算法:
- 初始化:随机选择K个数据点作为初始的聚类中心。
- 分配步骤:对于数据集中的每个点,根据其到每个聚类中心的距离,将其分配给最近的聚类中心。
- 更新步骤:对于每个聚类,计算所有分配给该聚类的数据点的平均值,然后将此平均值设置为新的聚类中心。
- 重复第2步和第3步,直到聚类中心不再变化。
EM算法在RNA-seq中的应用:
- 初始化:随机为多次映射的读取分配转录本来源,或使用其他方法提供初步估计。
- 预期步骤(E步骤):使用当前的估计来计算每个多次映射的读取最可能来自哪个转录本。
- 最大化步骤(M步骤):更新参数(例如转录本的丰度估计),使多次映射的读取来自这些转录本的似然度最大化。
- 重复第2步和第3步,直到参数估计收敛。
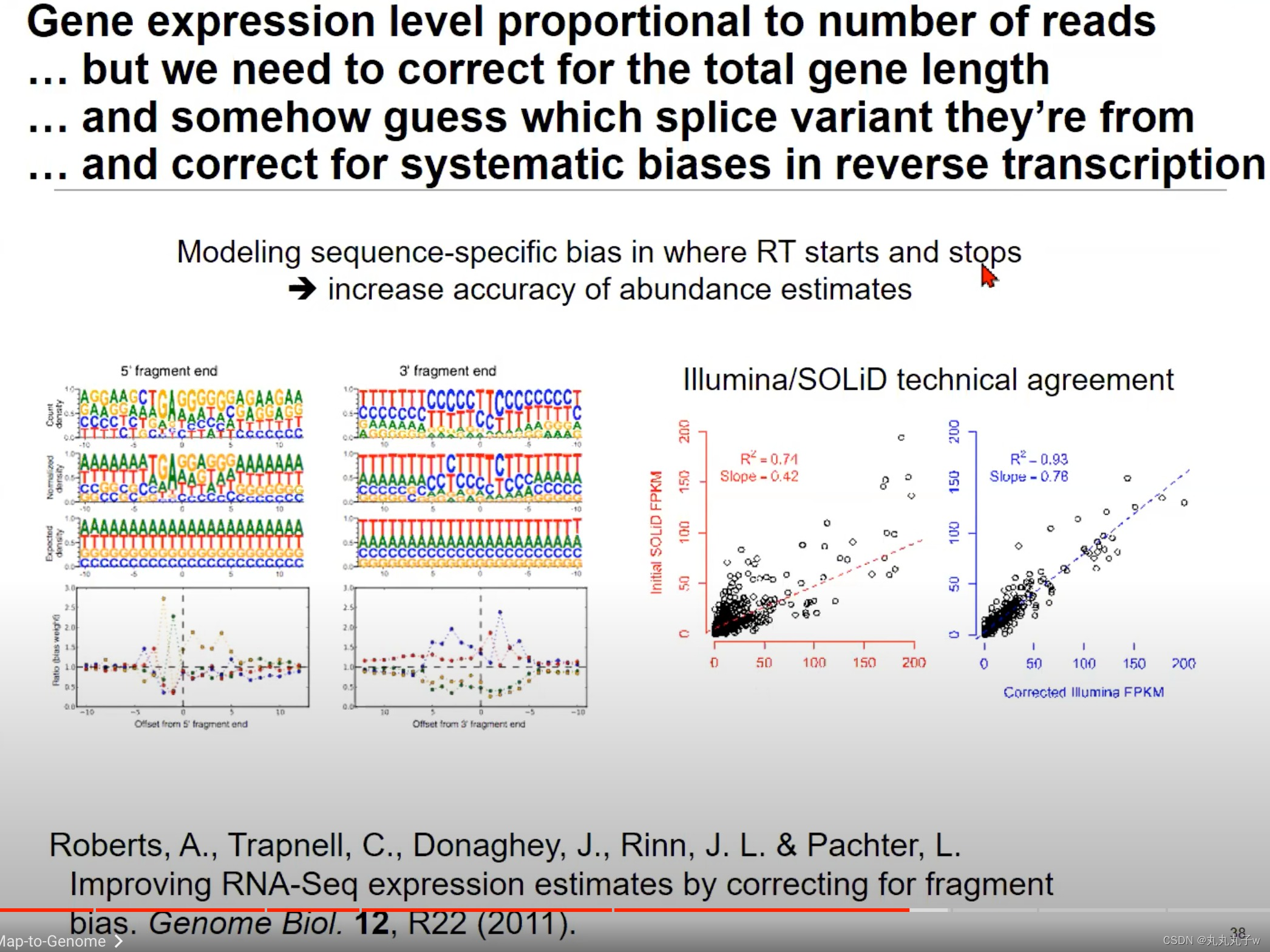
在RNA-Seq的过程中,反转录过程(RNA->cDNA)可能会引入偏差,需要进行纠正
- Modeling sequence-specific bias in where RT starts and stops:这部分强调了反转录过程在开始和结束时可能存在的序列特异性偏见。上方的两个图展示了在5’片段末端和3’片段末端处的一些核苷酸偏好。例如,在5’片段末端,G(鸟嘌呤)的出现频率很高。
- Illumina/SOLiD technical agreement:Illumina和SOLiD两种测序平台的技术对比。
- 图中点的分布表示两种技术在相同的样本中产生的FPKM值的对比。R^2值表示线性拟合的决定系数,而Slope表示斜率。理想情况下,所有点应该在y=x线上,表示两种技术产生的结果一致。
- 从图中可以看出,经过纠正后的Illumina数据与SOLiD数据有很好的一致性。

生物信息学100个基础问题 —— 第35题 RNA-Seq 数据的定量之RPKM和FPKM
FPKM:每千碱基转录本中每百万映射读取的片段数。RNA-Seq分析中常用的一种标准化方法,它可以帮助研究者更公平地比较不同样本和基因的表达水平。
FPKM如何工作:
- Per Kilobase(每千碱基):考虑到转录本的长度。例如,一个5kb的基因和一个1kb的基因,如果它们都有1000个读取,那么我们可以说长基因的表达是低的,因为读取是分布在一个更长的区域。
- Per Million mapped reads(每百万映射读取):这是为了标准化测序的深度。例如,一个样本有1000万的读取,另一个样本有5000万的读取,直接比较它们的读取数是不公平的。
3 Align-de-novo approaches
当我们没有参考基因组的时候该怎么办呢,比如还没有其完整基因组序列的生物、非模式生物
-
目标:
-
expressed gene content:识别在特定条件下表达的基因。
-
transcript abundance:测定不同转录本的丰度或水平。
-
differential expression:比较不同条件或处理下基因的表达差异。
-
3.1 Step 1: Generate all k-mers and link them together
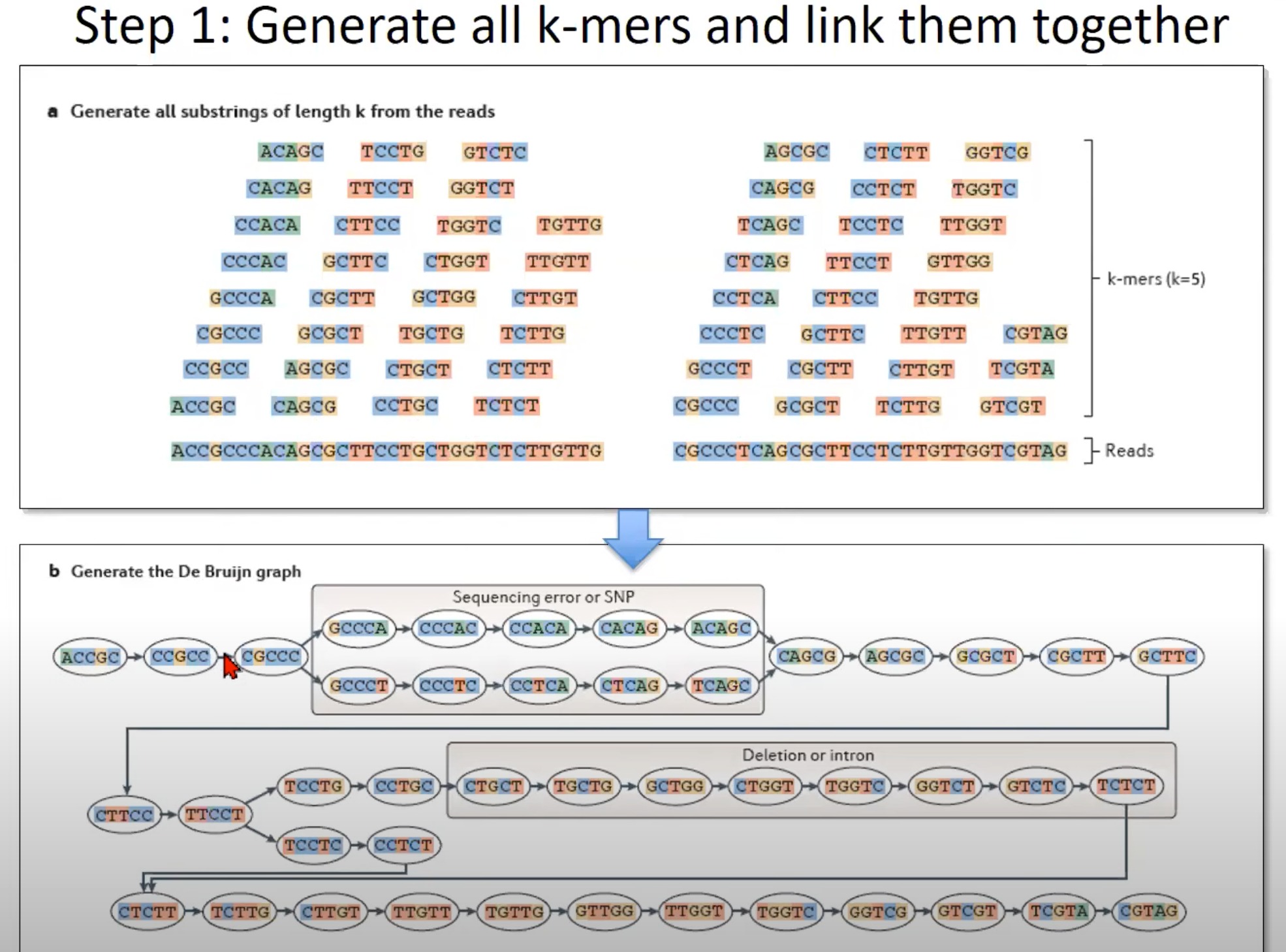
从RNA-Seq或DNA测序数据中生成所有的k-mer,并利用这些k-mer构建一个De Bruijn图
a. 生成所有长度为k的reads的子字符串
- 这里的示例使用k=5,这意味着从每个read中生成所有长度为5的子字符串。如图所示,从一个长的read中可以生成多个5-mer。
b. 生成De Bruijn图
- 根据k-mer生成De Bruijn图。
- 在De Bruijn图中,每个节点代表一个k-mer。例如,
GCCCA,CCCAC等。 - 有向边表示两个k-mer之间的关系。如果一个k-mer的后缀(除了第一个字符)与另一个k-mer的前缀(除了最后一个字符)相匹配,则在这两个k-mer之间存在有向边。
特定的标记:
-
Sequencing error or SNP:有些节点和边可能由于测序错误或单核苷酸多态性(SNP)而出现。这可能导致图中的额外路径。
-
Deletion or intron:在某些情况下,由于缺失或内含子的存在,可能会在图中观察到断裂。
-
我的一个疑惑:为什么不同的剪切外显子、剪切体不会导致这里误判检测出内含子吗
-
De Bruijn图是一种数据结构,用于de novo组装的方法。通过从测序reads中提取k-mer并在这些k-mer之间建立关系,可以重建原始的序列
一些意义:
- 简化组装过程:通过使用k-mer,生物信息学家可以将组装问题从复杂的对齐任务简化为图论问题。在De Bruijn图中,k-mer代表节点,而它们之间的连接代表可能的顺序关系。这种方法允许科研人员使用经典的图算法来解决组装问题。
- 克服测序误差:当处理数百万到数十亿的测序reads时,存在许多可能的测序误差。通过将reads转换为k-mer并在图中连接它们,可以识别并纠正这些误差,从而获得更准确的组装结果。
- 处理重复序列:许多基因组包含重复序列,这使得组装变得困难。使用k-mer和De Bruijn图可以更好地处理这些重复区域,因为它们可以明确地表示在图中的重复路径。
3.2 Step 2: Collapse the De Bruijn Graph
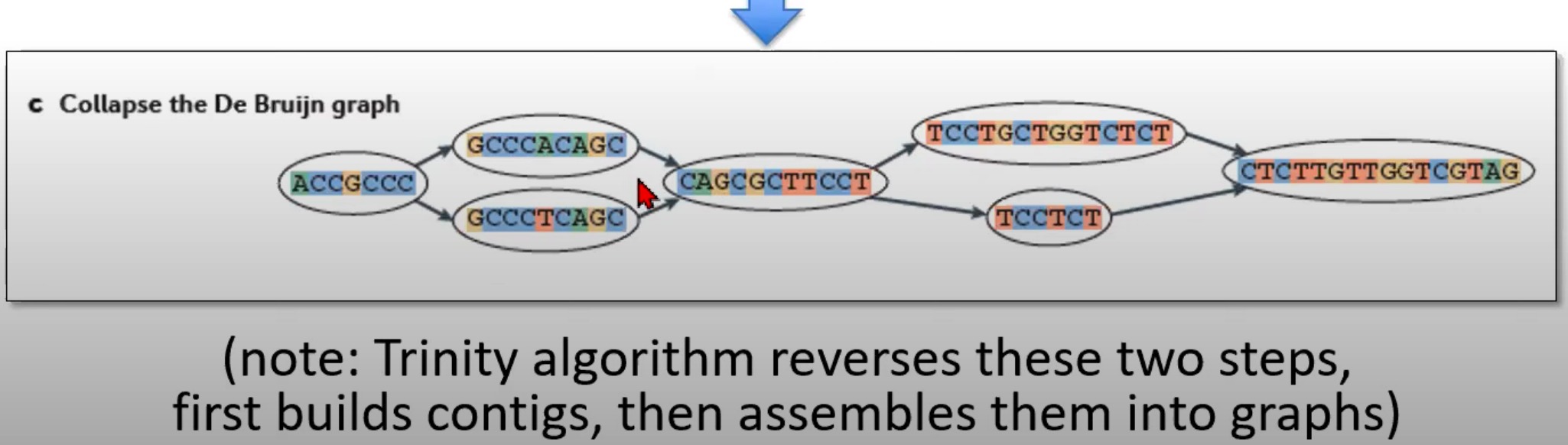
对De Bruijn图进行折叠来生成contigs
- 到由各种k-mer组成的De Bruijn图被合并或"折叠"为更长的序列,这些更长的序列称为contigs
- 折叠图的目的是简化这个图,合并连续且无分支的k-mers。
Trinity是一个专门为RNA-Seq数据设计的de novo转录组组装程序。与其他组装策略相反,Trinity首先构建contigs,然后将它们组装成图
3.3 Step 3: Resolve graph, quantify isoform abundance
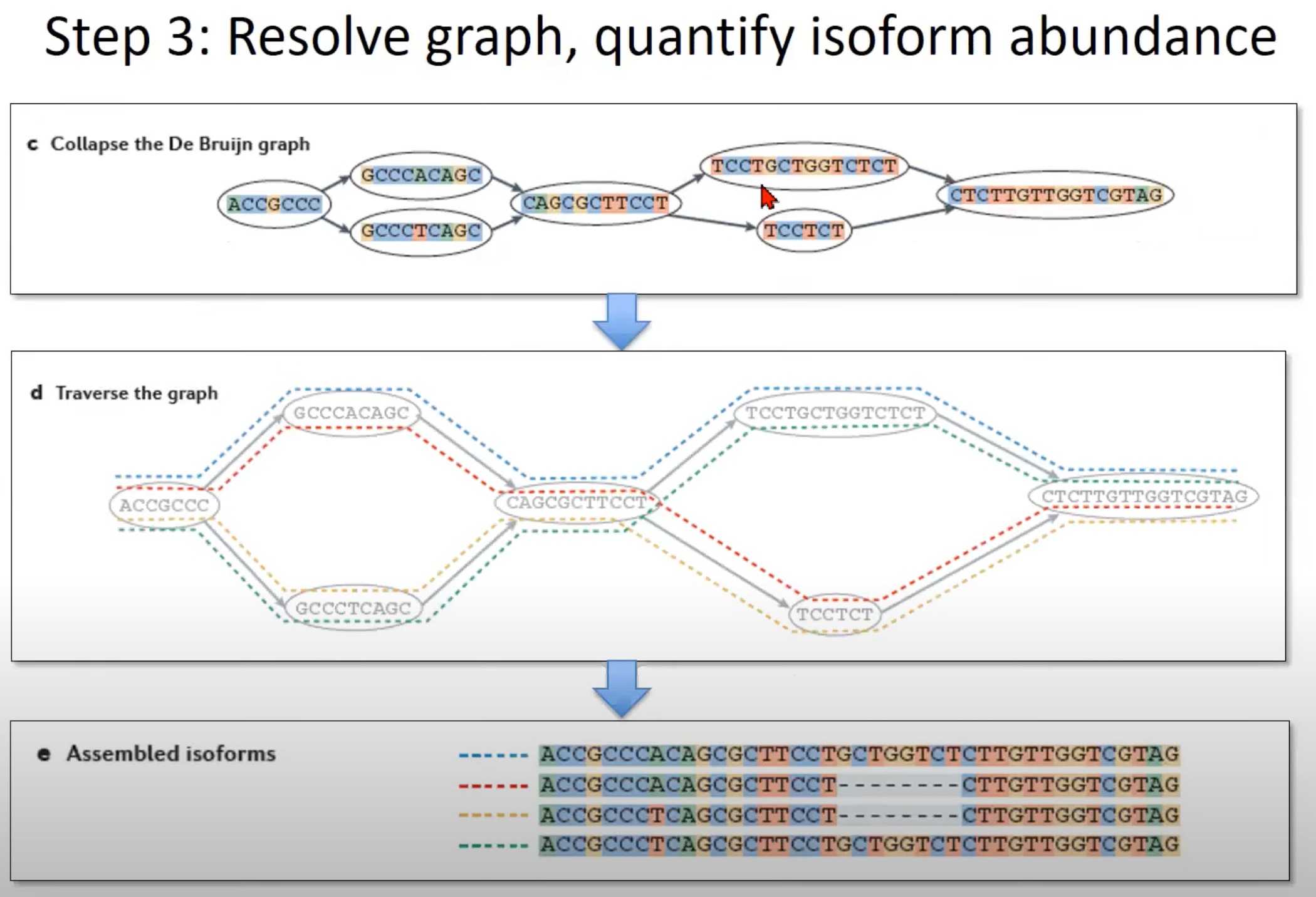
- 遍历图 (Step d):
- 为了重建可能的异构体,我们需要遍历图。这个图中展示了如何遍历,每种颜色代表一个可能的路径。
- 每条路径都代表一个可能的mRNA异构体。
- 组装的异构体 (Step e):
- 这一部分展示了遍历图后得到的mRNA异构体。在这里,我们可以看到三个可能的异构体。
- 通过这种方法,我们可以捕获一个基因由于选择性剪切产生的不同mRNA异构体。
- 最后还是需要计算一下异构体丰度。具体来说,将原始的测序reads比对回到组装得到的异构体上,根据比对结果统计覆盖度来估计每个异构体的丰度。
- Reads比对
- 计算覆盖度:对于每一个异构体,计算它的每一个位置上的reads覆盖度。
- 归一化:由于不同的异构体长度不同,直接使用覆盖度可能会导致长的异构体比短的异构体具有更高的估计丰度。因此,通常会对覆盖度进行归一化处理,得到每个异构体的RPKM或FPKM值。这两个值都考虑了异构体的长度和总的测序reads数目。
- 考虑多次比对的reads:一些reads可能比对到多个异构体,这样的reads需要特殊处理。一个常见的方法是按比对的质量将reads分配到不同的异构体。
- 用软件估计丰度:有一些专门的软件,如Cufflinks中的Cuffdiff和RSEM,可以估计RNA-seq数据中的转录本丰度。
- TPM归一化:近年来,TPM(Transcripts Per Million)成为了一个更为流行的归一化方法,因为它在样本之间更具有可比性。
4 Differential expression analysis

在进行RNA测序数据的分析时,我们先采用基于基因组的方法,如Tuxedo套件和Cufflinks,这些工具可以帮助我们处理单个的reads、拼接的reads以及评估转录本的丰度。
其次,当参考基因组不可用时或者你想从头组装,可以采用de novo组装方法。例如,Trinity工具可以帮助我们生成线性的contigs,将相关的contigs分组,以及确定可能的转录异构体。
至此,我们已经将reads比对到了refs上,统计出refs上每个gene对应的read counts。
通过比对control和treatment的read count。这时候就只要差异表达分析即可,在筛选出差异基因后,所谓的富集分析,常见的有GO,KEGG。这里就不细讲了。
我们即将进入差异表达分析。这一步是在处理RNA-seq数据时非常关键的,因为我们的目的通常是找出在不同条件或处理下表达有显著差异的基因或转录本。在这一阶段,我们会考虑多种变异性,如技术、生物以及不确定性。这些变异性因素可能会影响我们的分析结果,因此需要仔细处理和校正。
差异表达分析可以揭示那些在疾病状态与正常状态、处理与对照组、不同发育阶段或其他生物学条件下有显著变化的基因。
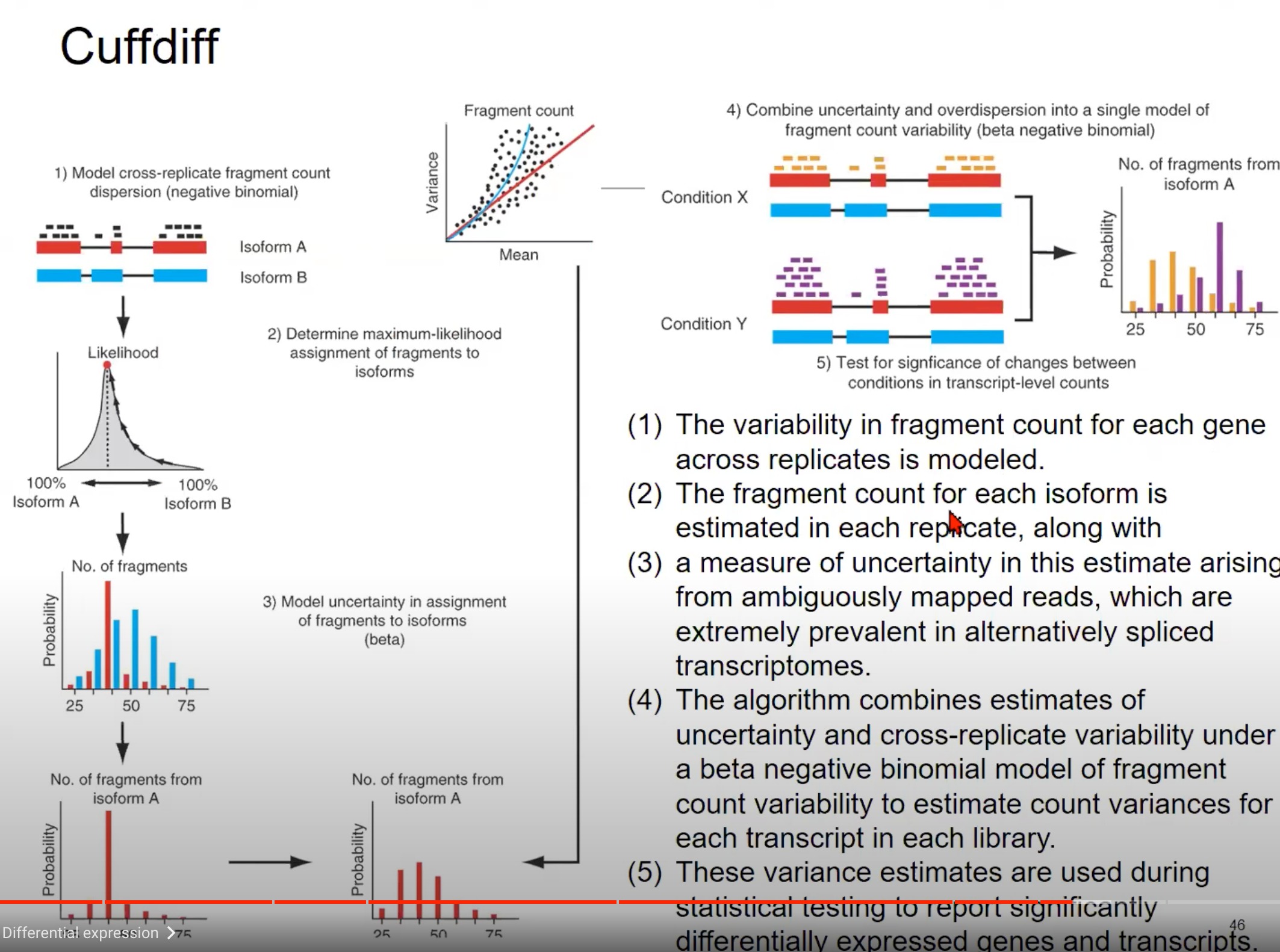
保持不确定性来进行估计
- 模型跨复制片段计数分散:图中展示了两种不同的转录本(Isoform A 和 Isoform B)的片段计数。这些片段计数在不同的复制品中会有所不同,这种变异性可以通过负二项分布来模型化。换句话说,由于生物学和技术的原因,每个基因的片段计数在不同的复制品中是有所不同的,我们使用负二项分布来捕获这种变异性。
- 确定片段的最大似然分配:对于给定的读取,我们需要确定它来自哪个转录本。这一步骤是通过计算读取分配给每个转录本的最大似然来实现的。如图中的似然图所示,我们可以估计一个读取来自Isoform A的概率和来自Isoform B的概率。
- 模型片段分配的不确定性:当读取可以映射到多个位置或多个转录本时,会产生不确定性。例如,由于选择性剪接,一个读取可能与多个转录本的一部分都匹配。这种不确定性可以使用beta分布来模型化。
- 合并不确定性和超分散:为了得到每个转录本的最终片段计数估计,算法结合了片段分配的不确定性(从第3步)和跨复制品的片段计数变异性(从第1步),使用beta负二项分布模型。
- 测试两种条件间的变化:最后,我们希望知道在不同条件(例如疾病状态和正常状态)下,转录本的表达是否发生了显著变化。为此,我们使用上述估计的方差来进行统计测试。
总结一下,这张图描述了如何使用Cuffdiff从RNA-seq数据中估计转录本的差异表达。它首先估计每个转录本的片段计数的变异性,然后结合这种变异性和片段分配的不确定性来得到最终的差异表达估计。

火山图!
这张图片展示了两种常用的绘图方法,用于分析和展示基因的差异表达数据:火山图 (Volcano plot) 和 MA图 (MA plot)。
- 火山图 (Volcano plot)
- 横轴 (x-axis):表示对数变化倍数 (Log2 fold change),它描述了基因在两个不同条件下的表达量之间的差异。
- 纵轴 (y-axis):表示p值的对数 (Log10 p-value),用于评估基因表达差异的统计学意义。
- 在此图中,图中红色的点代表显著差异表达的基因。这些点要么在正方向有很大的折叠变化(例如,大于某个阈值,如2倍),要么在负方向有很大的折叠变化(例如,小于某个阈值,如-2倍)。同时,它们的p值也非常小,表示这些差异是显著的。
- 火山图的名字来源于其形状,中间的部分看起来像火山的喷发部分,而两边上升的部分看起来像火山的坡。
- MA图 (MA plot)
- 横轴 (x-axis):表示基因的平均表达水平 (Log2 Average Expression level)。
- 纵轴 (y-axis):表示基因在两个条件之间的对数变化倍数 (Log2 fold change)。
- MA图提供了关于基因表达差异与其平均表达水平之间关系的视觉表示。红色的点代表在给定的平均表达水平下显著差异表达的基因。
- “M” 是指差异 (表示为对数变化倍数),而"A" 是指平均强度 (表示为平均表达水平)。
此外,图中还指出了“显著差异表达的转录本具有FDR <= 0.001”。FDR是“假发现率”(False Discovery Rate)的缩写,是一种在多重假设检验中控制误差的方法。当FDR非常小(例如,小于或等于0.001)时,这意味着差异表达结果的可靠性非常高。

![2023年中国调速器产量、销量及市场规模分析[图]](https://img-blog.csdnimg.cn/img_convert/2e6d70d002f6a1092e439123f22dc749.png)