文章目录
- biref
- 统计学原理
- 其他注意事项
- 代码实现部分
biref
前情提要链接:
https://blog.csdn.net/jiangshandaiyou/article/details/136536349
https://blog.csdn.net/jiangshandaiyou/article/details/134457515
相比起GSA,GSEA不再关注于差异基因,因此不受p-value以及log2FC的指标的过滤影响,可以获得更多生物学功能变化的信息。但是呢需要多个样本的测序数据才能得到与表型相关的排序数据,如果我只有一个样本呢?或者我的样本没有先验的表型区分呢(也就是没法分组)?
ssGSEA说我可以。ssGSEA是单样本的GSEA,对于一个基因集S,每一个样本都可以计算得到一个enrichment score。
也就是说其实我们可以从一个多样本的表达矩阵得到另一个矩阵,行是多个不同的基因集S,列仍然是样本,值则是ES。
Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462(7269):108-112. doi:10.1038/nature08460
统计学原理
需要补充的基础知识:
https://blog.csdn.net/jiangshandaiyou/article/details/136545010
其他注意事项
- GSEA与ssGSEA几乎一模一样,它们都需要对所有基因进行排序得到一个ranked gene list ;
都运用了k-s like test评估基因集S中的基因与S外的基因的分布是否不同,
虽然是思想是一模一样的,但是统计量的计算不同。 - 在GSEA中,基因的是按照log2FC从大到小排列,而在ssGSEA中,对于单个样本,将基因按照其表达量从大到小排序。也就是每个样本都是一个独特他的ranked gene list。
- K-s like test中的eCDF阶梯上升高度所依赖的值不同。
在GSEA中,最后基因集S中基因的阶梯上升高度依赖于log2FC的加权值。
而在ssGSEA中,为了消除异常值对结果的影响,在第一步排序基因后,会将表达量用秩次(rank)替换。
比如说一个样本有100个基因的信息,先按照基因表达量排好序后,这个genelist对应的值是基因的表达量,然后经过秩次标准化转换后,这个genelist已经由表达量的降序排列变成了100,99,98,…,3,2,1。
也就是原文L中的r1,r2 ,r3 ,…,rN。所以最后基因集S中的基因的阶梯上升高度依赖于秩次的加权值。

代码实现部分
# 借助GSVA工具实现
# .libPaths("C:/Users/lenovo/Documents/R/win-library/4.0")
library(tidyverse)
library(reshape2)
library(cowplot)
library(RColorBrewer)
library(GSVA)options(warn = -1)
options(digits = 5)# 准备表达矩阵
list.files("G:/20240223-project-HY0007-GSVA-analysis-result/")expr <- read.table("../TPM_DE.filter.txt",sep = "\t",header = T,row.names = 1)
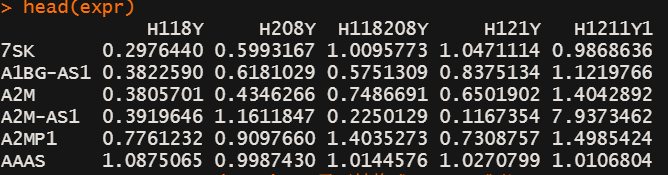
head(expr)
expr <- as.matrix(expr) # 需要转换成matrix或者 ExpressionSet object# 准备预设的gene sets
# install.packages("msigdbr")
library(msigdbr)
## msigdbr包提取下载 先试试KEGG和GO做GSVA分析
##KEGG
KEGG_df_all <- msigdbr(species = "Homo sapiens", # Homo sapiens or Mus musculuscategory = "C2",subcategory = "CP:KEGG")
KEGG_df <- dplyr::select(KEGG_df_all,gs_name,gs_exact_source,gene_symbol)
kegg_list <- split(KEGG_df$gene_symbol, KEGG_df$gs_name) ##按照gs_name给gene_symbol分组##GO
GO_df_all <- msigdbr(species = "Homo sapiens",category = "C5")
GO_df <- dplyr::select(GO_df_all, gs_name, gene_symbol, gs_exact_source, gs_subcat)
GO_df <- GO_df[GO_df$gs_subcat!="HPO",]
go_list <- split(GO_df$gene_symbol, GO_df$gs_name) ##按照gs_name给gene_symbol分组#### GSVA ####
# geneset 1
geneset <- go_list
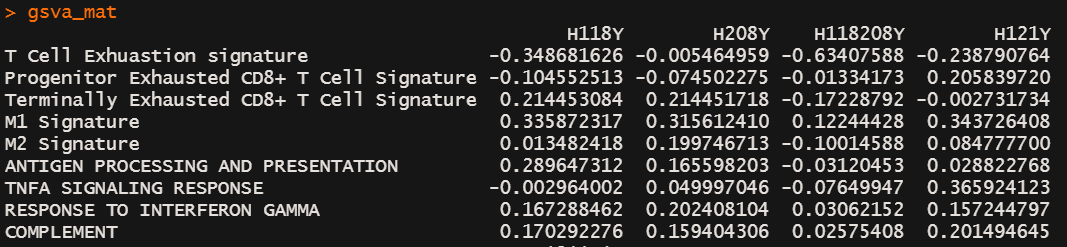
gsva_mat <- gsva(expr=expr, gset.idx.list=geneset, kcdf="Gaussian" ,#"Gaussian" for logCPM,logRPKM,logTPM, "Poisson" for countsverbose=T, mx.diff =TRUE,# 下游做limma得到差异通路min.sz = 10, # gene sets 少于10个gene的过滤掉method = "ssgsea"# method=c("gsva", "ssgsea", "zscore", "plage"))write.csv(gsva_mat,"gsva_go_matrix.csv")# geneset 2
geneset <- kegg_list
gsva_mat <- gsva(expr=expr, gset.idx.list=geneset, kcdf="Gaussian" ,#"Gaussian" for logCPM,logRPKM,logTPM, "Poisson" for countsverbose=T, mx.diff =TRUE,# 下游做limma得到差异通路min.sz = 10, # gene sets 少于10个gene的过滤掉method = "ssgsea"# method=c("gsva", "ssgsea", "zscore", "plage")
)write.csv(gsva_mat,"gsva_kegg_matrix.csv")