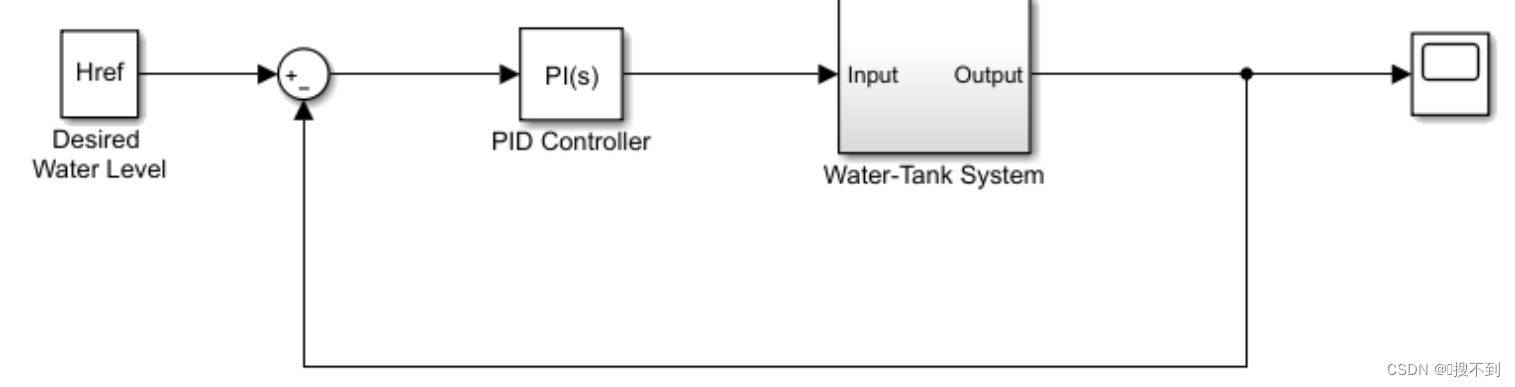
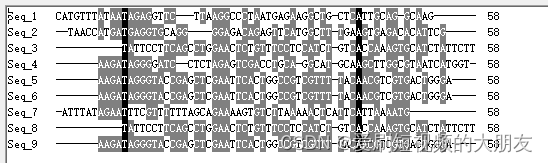
观察数据
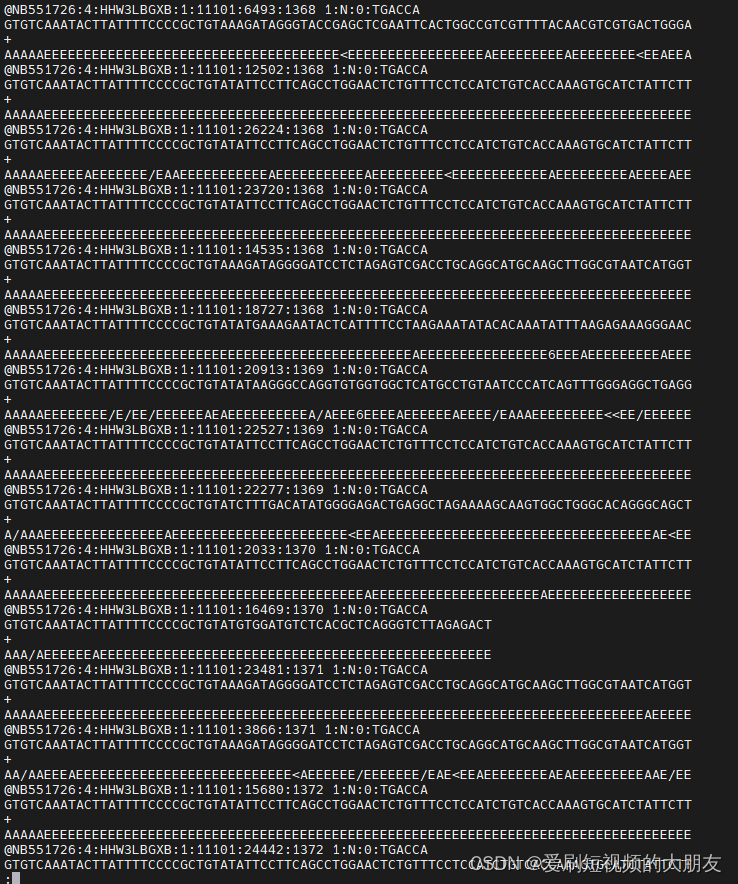
这是经公司使用fastp质控后的数据,我们先挑选部分数据进行比对,观察序列结构
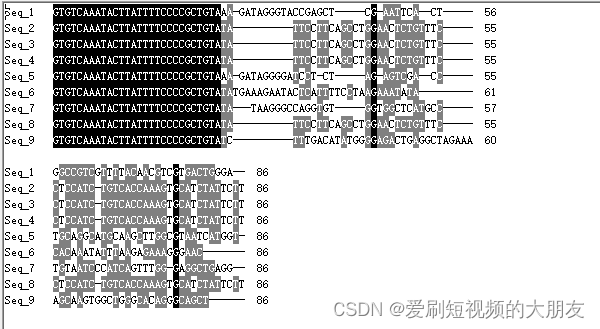
为了准确性,我们再次挑选另一批数据进行比对
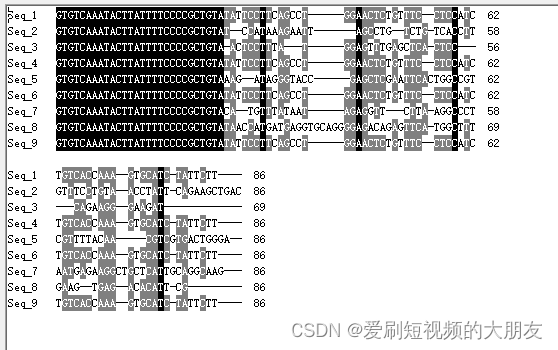
可以看到,所有序列都存在一个“GTGTCAAATACTTATTTTCCCCGCTGTA”的前导序列,这可能是接头序列之类的,我们使用cutadapt工具将其去除。
nohup cutadapt -g GTGTCAAATACTTATTTTCCCCGCTGTA -o ps_fastpTrimmed.cutadapt.fq PS_fastpTrimmed.fastq &
运行日志
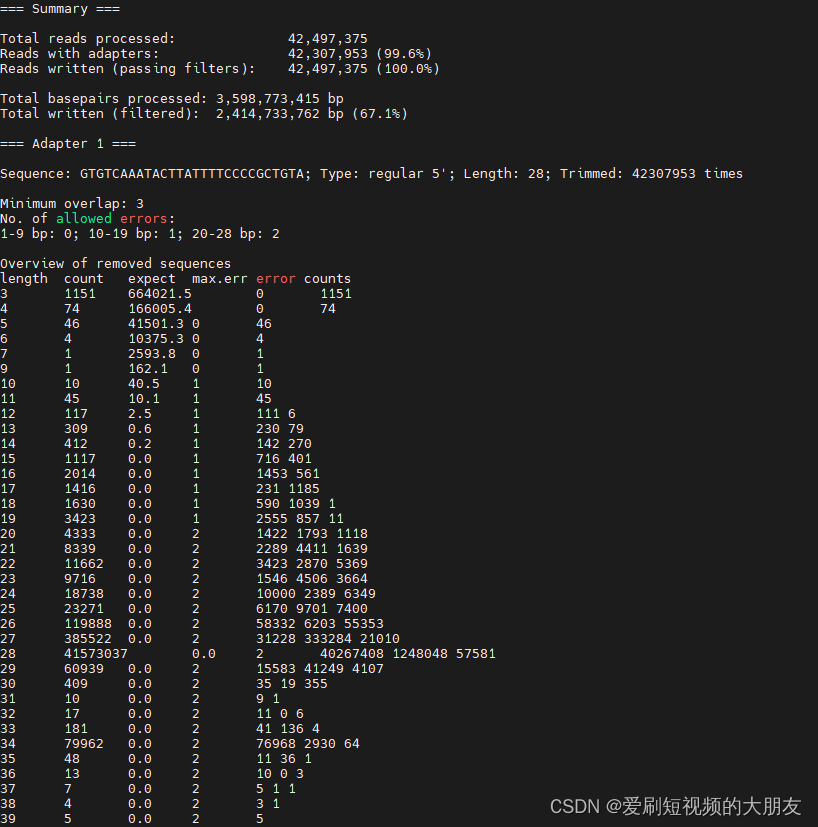 运行结果
运行结果
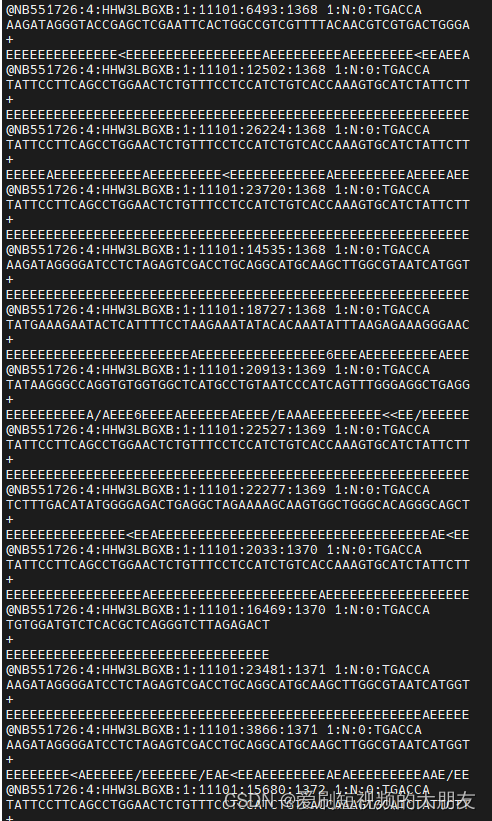
所有数据的该序列都被去除,我们再来比对一下,看看是否存在还需要修剪的序列。
比对
bwa mem -t 32 ../001.blastFORcgspy/human.ref.bwaindex/GCF_000001405.40_GRCh38.p14_genomic.fna ps_fastpTrimmed.cutadapt.fq > ps_fastpTrimmed.cutadapt.sam &
运行日志
[M::bwa_idx_load_from_disk] read 0 ALT contigs ✔ ⚙ 911 13:15:02
[M::process] read 5632178 sequences (320000099 bp)...
[M::process] read 5632870 sequences (320000113 bp)...
[M::mem_process_seqs] Processed 5632178 reads in 574.971 CPU sec, 19.185 real sec
[M::process] read 5630606 sequences (320000003 bp)...
[M::mem_process_seqs] Processed 5632870 reads in 552.172 CPU sec, 17.734 real sec
[M::process] read 5632256 sequences (320000078 bp)...
[M::mem_process_seqs] Processed 5630606 reads in 650.468 CPU sec, 20.646 real sec
[M::process] read 5631510 sequences (320000045 bp)...
[M::mem_process_seqs] Processed 5632256 reads in 641.476 CPU sec, 20.295 real sec
[M::process] read 5631304 sequences (320000059 bp)...
[M::mem_process_seqs] Processed 5631510 reads in 645.206 CPU sec, 20.259 real sec
[M::process] read 5631206 sequences (320000009 bp)...
[M::mem_process_seqs] Processed 5631304 reads in 591.624 CPU sec, 18.888 real sec
[M::process] read 3075445 sequences (174733356 bp)...
[M::mem_process_seqs] Processed 5631206 reads in 594.674 CPU sec, 18.734 real sec
[M::mem_process_seqs] Processed 3075445 reads in 384.189 CPU sec, 12.525 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 32 ../001.blastFORcgspy/human.ref.bwaindex/GCF_000001405.40_GRCh38.p14_genomic.fna ps_fastpTrimmed.cutadapt.fq
[main] Real time: 168.989 sec; CPU: 4645.033 sec去除表头
awk '!/@SQ/' ps_fastpTrimmed.cutadapt.sam > ps_fastpTrimmed.cutadapt.1.sam
查看比对结果
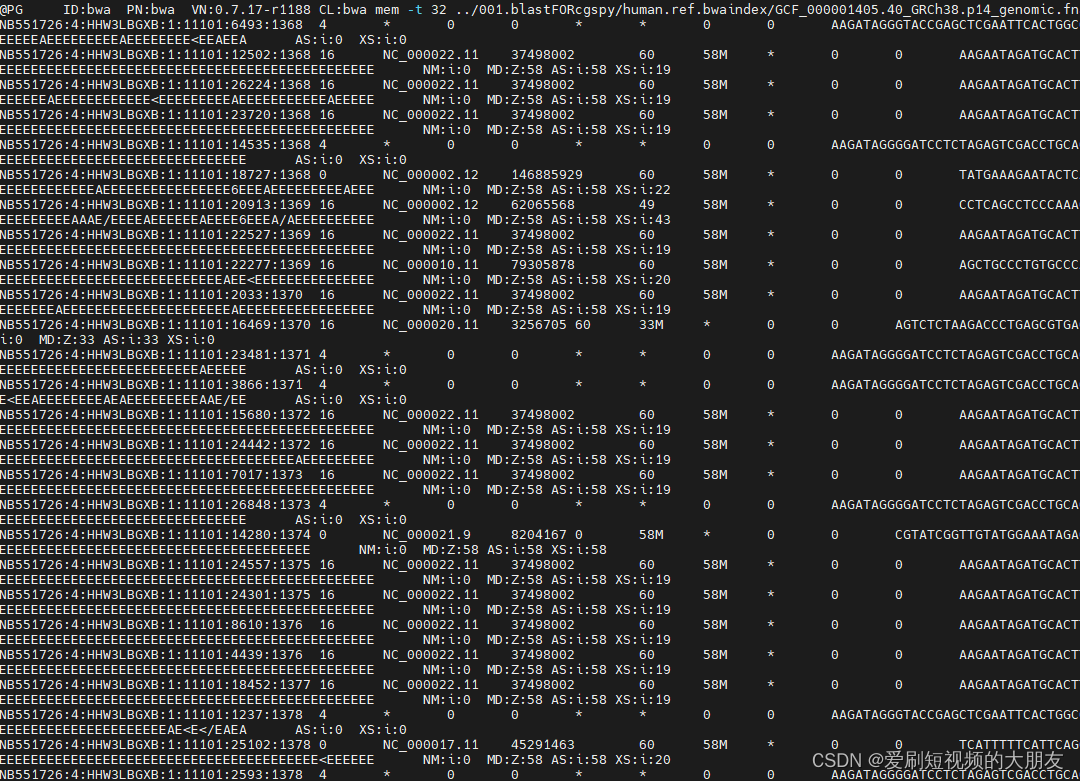 提取文件的前六列
提取文件的前六列
awk '{print $1, $2, $3, $4, $5, $6}' ps_fastpTrimmed.cutadapt.1.sam > ps_fastpTrimmed.cutadapt.2.sam &
去除没有匹配上的数据
awk '$4 != 0' ps_fastpTrimmed.cutadapt.2.sam > ps_fastpTrimmed.cutadapt.3.sam &
提取文件的2,3,4列(由于文件太大不方便excel统计,尝试一下)
awk '{print $2, $3, $4}' ps_fastpTrimmed.cutadapt.3.sam > ps_fastpTrimmed.cutadapt.4.sam &
不行,还是太大了,文件超过了三千万行,远超过了excel的处理能力,寻找其他方法进行统计。
使用samtools统计每个位点覆盖到的reads数量。
首先使用samtools将未经处理的比对结果转换为bam文件
samtools view -bS input.sam > output.bam
使用samtools软件对bam文件进行索引
(注意:在使用samtools对bam文件进行索引之前必须对bam文件进行排序,否则会报错)
samtools sort ps_fastpTrimmed.cutadapt.bam -o ps_fastpTrimmed.cutadapt.sorted.bam &
samtools index ps_fastpTrimmed.cutadapt.sorted.bam &
然后,使用samtools depth命令统计每个位点的覆盖 reads 数:
samtools depth alignment.bam > coverage.txt