可以使用以下命令来查看 gz 压缩文件的内容: zcat file.gz 1 该命令会将 file.gz 文件解压并输出到标准输出,可以通过管道符将其与 grep 命令结合使用来查找需要的关键词,例如: zcat file.gz | grep keyword 1 该命令会将 file.gz 文件解压并输出到标准输出,然后通过管道符将其传递给 grep 命令,查找包含关键词 “keyword” 的行。
挖掘公共单细胞数据集时,会遇到常见各种单细胞测序数据格式。现总结如下,方便自己日后调用,以创建Seurat对象
(1)barcodes.tsv.gz、features.tsv.gz、matrix.mtx.gz
(2)表达矩阵
(3)h5
(4)h5ad
格式一:barcodes.tsv.gz、features.tsv.gz、matrix.mtx.gz【☆】
- 这是cellranger上游比对分析产生的3个文件,分别代表细胞标签(barcode)、基因ID(feature)、表达数据(matrix)
- 一般先使用
read10X()对这三个文件进行整合,得到行为基因、列为细胞的表达矩阵(为稀疏矩阵dgCMatrix格式,节约内存);然后再配合CreateSeuratObject()函数创建Seurat对象 - 示例数据集:GSE166635,创建代码如下----

https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE166635
dir="./data/HCC2/filtered_feature_bc_matrix/"
list.files(dir)
#[1] "barcodes.tsv.gz" "features.tsv.gz" "matrix.mtx.gz" counts <- Read10X(data.dir = dir)
class(counts)
#[1] "dgCMatrix"
#attr(,"package")
#[1] "Matrix"scRNA <- CreateSeuratObject(counts = counts)
scRNA
#An object of class Seurat
#33694 features across 9112 samples within 1 assay
#Active assay: RNA (33694 features, 0 variable features)
- 如上
Read10X()函数接受的参数为目录名,该目录包含了所需的三个配套文件;值得注意的是三个文件名只能分别是barcodes.tsv.gz、features.tsv.gz、matrix.mtx.gz,然后read10X函数可以自动加载。如上截图那样就是需要修改的~
关于barcodes.tsv.gz、features.tsv.gz、matrix.mtx.gz三个文件的格式与内容
- 一般来说直接使用
read10X()不会出现什么问题,但今天遇到GSE148192数据集时,出现了报错~~
dir = "./GSE148192_RAW/GSM4462451/"
list.files(dir)
#[1] "barcodes.tsv.gz" "features.tsv.gz" "matrix.mtx.gz"
counts = Read10X(dir)
#Error in dimnamesGets(x, value) :
# invalid dimnames given for “dgTMatrix” object
- 所以这个GSE ID提供的数据格式可能是有点问题,接下来就通过对比GSE166635的GSM5076750(可以正常读入)与GSE148192的GSM4462451(读入失败),探索下这三个文件的格式
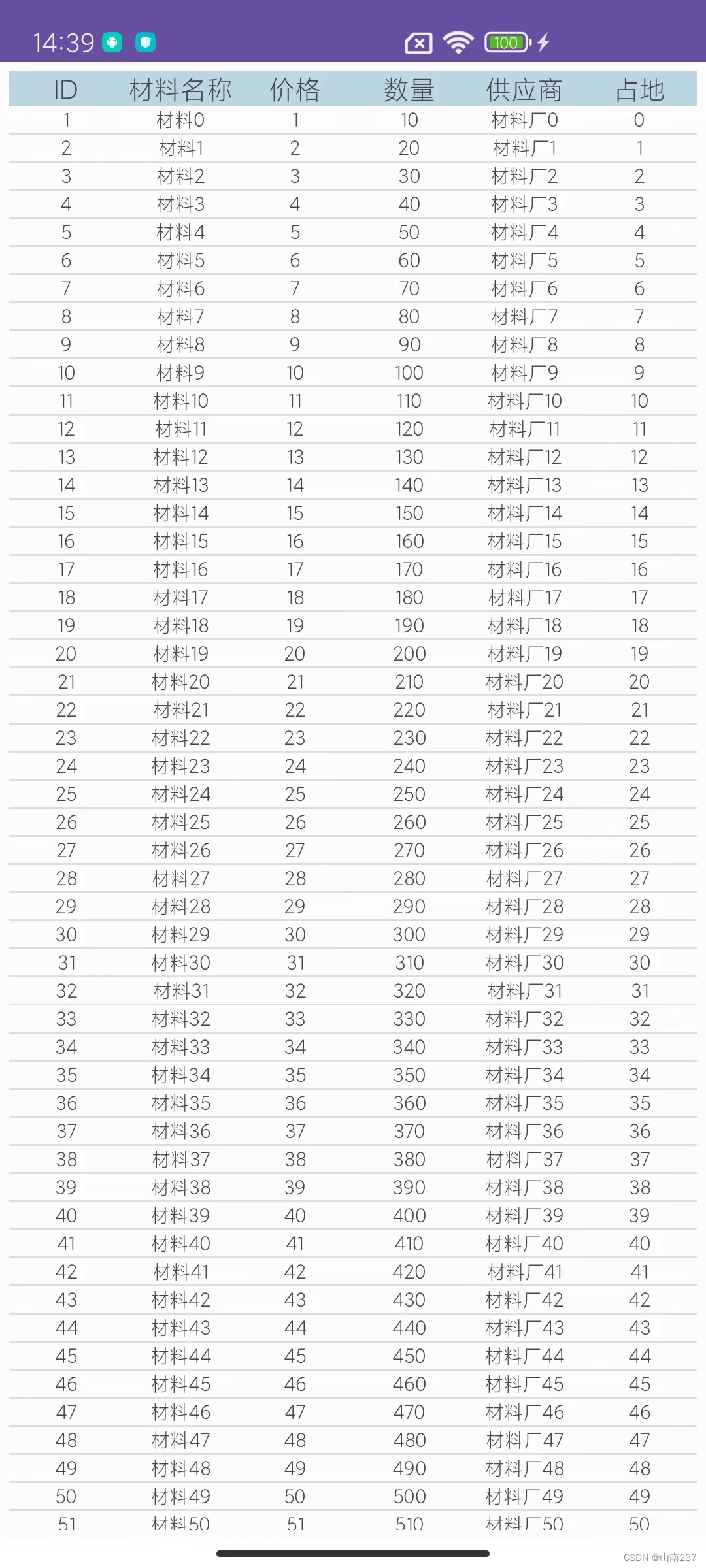
(1)barcodes.tsv.gz
-
GSM5076750的格式:如下看出就简单的一列,为细胞的barcode标签信息

-
GSM4462451的格式:如下看出,区别在于多了行名,以及三列细胞注释信息



(2)features.tsv.gz
-
GSM5076750的格式:如下可以看出均为基因的注释信息,前两列为基因ID

-
GSM4462451的格式:如下看出,区别在于同样多了行名,以及额外两列信息



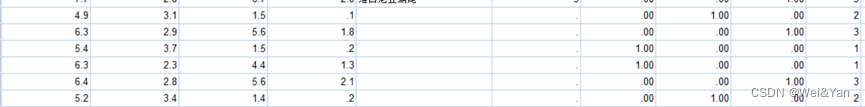
(3)matrix.mtx.gz
- GSM5076750的格式:如下(前三行为注释信息,其中第三行为total number genes、cells、counts),结合上述细胞标签与基因名信息,知道了前两列分别为基因和细胞的索引,第三列为表达信息。
利用这种方式实现了高效的储存数据(值得借鉴学习)。以第四行为例:表示barcodes.tsv.gz文件里第一个细胞的features.tsv.gz第33665个基因的counts数为22。
-
GSM4462451的格式:如下看出,区别有两点:第一列为细胞索引、第二列为基因索引,并且第3列是非整型数据。



经过一番探索,将GSM4462451的
barcodes.tsv.gz、features.tsv.gz行名删除;matrix.mtx.gz的第一列与第二列调换,第三列改为整型后,read10X()便可以顺利都成功。我认为GSM4462451这几个文件应该是作者自己制作的,吐槽一下~~。不过了解了一番这三个文件的格式也是有所收获。

格式二:直接提供表达矩阵
- 这种是最方便的,直接创建Seurat即可
- 示例数据:GSE144320

scRNA <- CreateSeuratObject(counts = counts)
scRNA
格式三:h5格式文件
- 使用
Read10X_h5()函数,读入表达矩阵,在创建Seurat对象 - 示例数据:GSE138433

image.png
sce <- Read10X_h5(filename = GSM4107899_LH16.3814_raw_gene_bc_matrices_h5.h5")
sce <- CreateSeuratObject(counts = sce)
格式四:h5ad格式
- 需要安装,使用
SeuratDisk包的两个函数; - 先将后
h5ad格式转换为h5seurat格式,再使用LoadH5Seurat()函数读取Seurat对象。 - 示例数据集:GSE153643
#remotes::install_github("mojaveazure/seurat-disk")
library(SeuratDisk)
Convert("GSE153643_RAW/GSM4648565_liver_raw_counts.h5ad", "h5seurat",overwrite = TRUE,assay = "RNA")
scRNA <- LoadH5Seurat("GSE153643_RAW/GSM4648565_liver_raw_counts.h5seurat")
#注意一下,我之前载入时,表达矩阵被转置了,需要处理一下~

以上是我目前了解到的针对不同数据来源,创建Seurat对象的几种方式。如遇新的方法,会继续补充~~
©著作权归作者所有,转载或内容合作请联系作者
作者:小贝学生信
链接:https://www.jianshu.com/p/5b26d7bc37b7
来源:简书
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。