检索到目标数据集后,开始数据挖掘,本文以阿尔兹海默症数据集GSE1297为例
目录
GEOquery 简介
安装并加载GEOquery包
getGEO函数获取数据(联网下载)
更换下载数据源
对数据集进行初步观察处理
GEOquery 简介
GEOquery是一个在生物信息学中常用的R语言包,用于从NCBI Gene Expression Omnibus(GEO)数据库中获取和分析基因表达数据。以下是GEOquery包的简介:
1. 数据获取:GEOquery包提供了方便的函数来从GEO数据库中获取基因表达数据。您可以使用`getGEO()`函数来下载和导入GEO数据集,包括微阵列和高通量测序数据。
2. 数据处理:GEOquery包提供了一系列函数来处理GEO数据集。您可以使用`pData()`函数获取样本的基本信息,使用`exprs()`函数获取基因表达矩阵,使用`featureNames()`函数获取基因名字等。
3. 数据质量控制:GEOquery包提供了一些函数来进行数据质量控制。您可以使用`boxplot()`和`plotDensities()`函数来检查数据的分布情况,使用`arrayQualityMetrics()`函数来评估数据的质量。
4. 数据分析:GEOquery包结合了其他常用的R语言包,如limma、edgeR等,提供了丰富的数据分析方法。您可以使用这些方法进行差异表达分析、聚类分析、富集分析等。
安装并加载GEOquery包
# 安装并加载GEOquery包if (!requireNamespace("GEOquery", quietly = TRUE))install.packages("GEOquery")
#BiocManager: : install("GEOquery")library(Biobase)
library(GEOquery)
GEOquery如果安装失败可以尝试
BiocManager: : install("GEOquery")getGEO函数获取数据(联网下载)
`getGEO`函数有一些可选参数,可以用来自定义数据获取和处理过程。以下是一些常用的参数介绍:
1. `GEO`:指定要获取的GEO数据集的ID。可以是一个字符向量,包含多个ID,用于同时获取多个数据集。
2. `destdir`:指定下载数据的目标文件夹。默认情况下,数据会下载到当前工作目录下的"geoquery"文件夹中。
3. `getGPL`:逻辑值,表示是否同时获取与GEO数据集关联的平台信息。默认为FALSE,不获取平台信息。
4. `AnnotGPL`:逻辑值,表示是否获取平台的注释信息。默认为FALSE,不获取注释信息。
5. `GSEMatrix`:逻辑值,表示是否将基因表达数据存储为GSEMatrix对象。默认为TRUE,将数据存储为GSEMatrix对象。
6. `getGPLcdf`:逻辑值,表示是否获取与GEO数据集关联的平台注释文件。默认为FALSE,不获取注释文件。
7. `verbose`:逻辑值,表示是否显示下载和导入过程的详细信息。默认为TRUE,显示详细信息。
# 指定GEO数据集的ID
gse_id <- "GSE1297"# 使用getGEO函数获取数据
gse_data <- getGEO(gse_id,destdir = ".", AnnotGPL = FALSE)更换下载数据源
数据集大时,国内网络经常会出现中断,需要更换下载源
chooseCRANmirror() #选择镜像源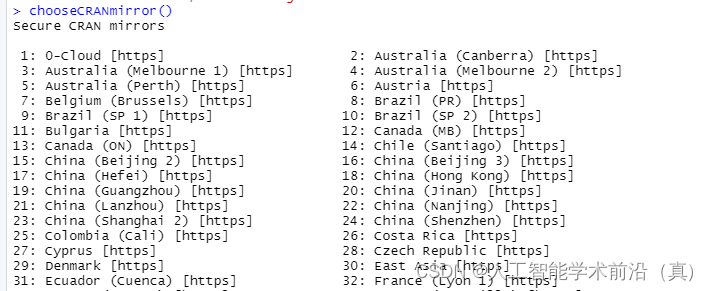
输入数字选择国内镜像源
在中国选择合适的CRAN镜像源可以提高下载速度和稳定性。以下是几个常用的CRAN镜像源:
1. 中国科学技术大学镜像源:https://mirrors.ustc.edu.cn/CRAN/
2. 清华大学镜像源:https://mirrors.tuna.tsinghua.edu.cn/CRAN/
3. 阿里云镜像源:https://mirrors.aliyun.com/CRAN/
您可以尝试使用其中一个镜像源。一般来说,中国科学技术大学镜像源和清华大学镜像源在中国使用较为广泛且速度较快。如果您发现某个镜像源下载速度较慢,可以尝试切换到其他镜像源进行下载。
对数据集进行初步观察处理
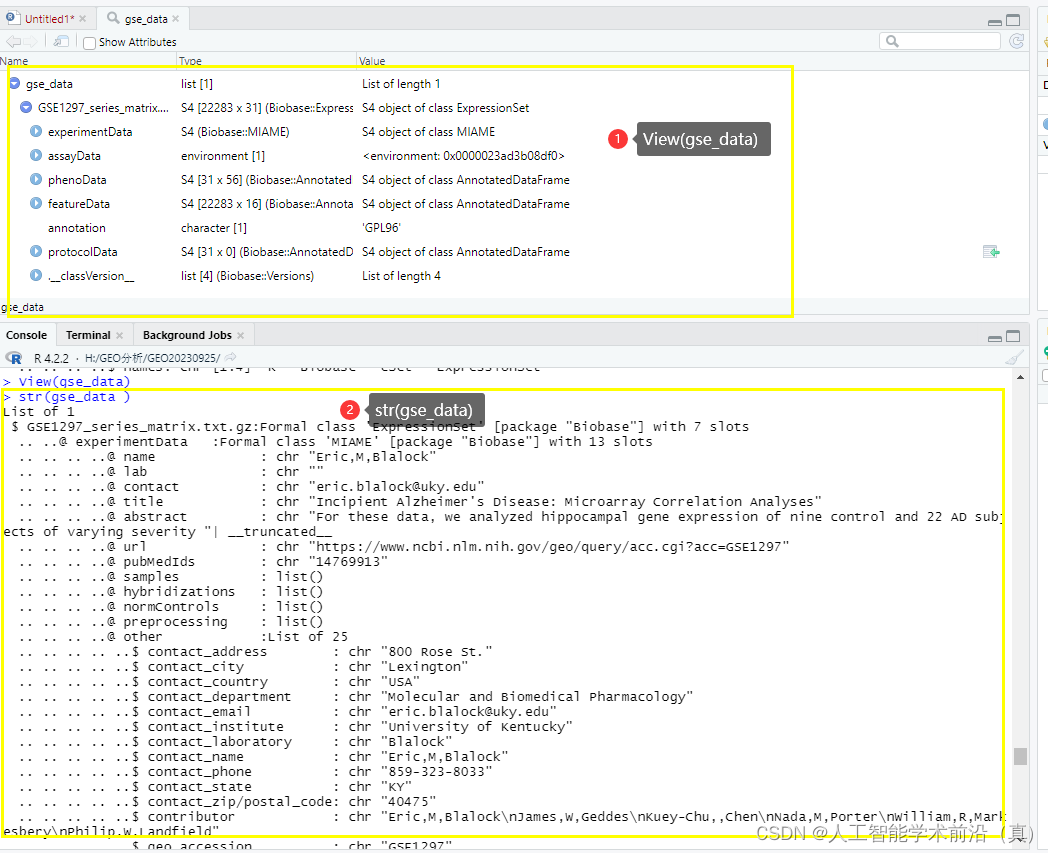
#查看对象结构信息
View(gse_data)
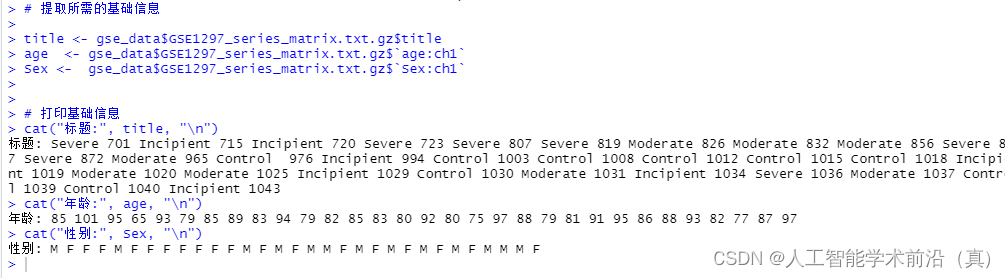
str(gse_data )# 提取所需的基础信息title <- gse_data$GSE1297_series_matrix.txt.gz$title
age <- gse_data$GSE1297_series_matrix.txt.gz$`age:ch1`
Sex <- gse_data$GSE1297_series_matrix.txt.gz$`Sex:ch1`# 打印基础信息
cat("标题:", title, "\n")
cat("年龄:", age, "\n")
cat("性别:", Sex, "\n")# 查看数据的摘要信息
summary(gse_data)# 提取所需的数据
expression_data <- exprs(gse_data[[1]])查看数据结构
根据需求提取基础信息
获取临床信息(后续工作分组需要)
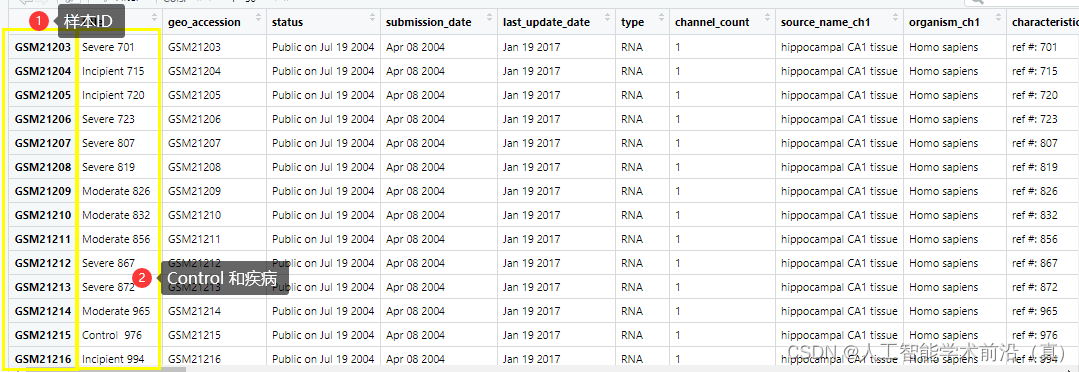
提取基因表达数据

所有代码
if (!requireNamespace("GEOquery", quietly = TRUE))install.packages("GEOquery")
#BiocManager: : install("GEOquery")#library(Biobase)
library(GEOquery)# 指定GEO数据集的ID
gse_id <- "GSE1297"
#标题: Gene expression patterns in human cancer cell lines
#摘要: This dataset contains gene expression data from various human cancer cell lines. The data was generated using microarray technology.
#平台: GPL570
#样本数量: 60chooseCRANmirror() #选择镜像源
# 使用getGEO函数获取数据
gse_info <- getGEO(gse_id,destdir = ".", AnnotGPL = FALSE)#查看对象结构信息
str(gse_info )# 提取所需的基础信息title <- gse_info$GSE1297_series_matrix.txt.gz$title
age <- gse_info$GSE1297_series_matrix.txt.gz$`age:ch1`
Sex <- gse_info$GSE1297_series_matrix.txt.gz$`Sex:ch1`# 打印基础信息
cat("标题:", title, "\n")
cat("年龄:", age, "\n")
cat("性别:", Sex, "\n")# 查看数据的摘要信息
summary(gse_info)
summary(gse_info$GSE1297_series_matrix.txt.gz)# 提取所需的数据
expression_data <- exprs(gse_info[[1]])#+========================================================================================# 样本编号方法一
samples =gse_info$GSE1297_series_matrix.txt.gz$geo_accession
# 样本编号方法二
samples=sampleNames(gse_info) # sample name就是看有多少GSM样本
# 样本编号方法三
samples=as.character(pdata[,2])#+========================================================================================#提取临床信息 方法一:$或者@ ,配合str()观察结构
pdata = gse_info$GSE1297_series_matrix.txt.gz@phenoData@data#提取临床信息 方法二:用函数提取 提取表达矩阵
phenoData= gse_info$GSE1297_series_matrix.txt.gz@phenoData
pdata = pData(phenoData)#方法三:直接通过鼠标操作再粘贴 在表达矩阵前面的注释信息里面#+========================================================================================# 提取基因表达矩阵
expression_data <- exprs(gse_info[[1]])#+========================================================================================
基因表达数据已经提取到了,但是,矩阵的行名还是芯片探针的名称。
下节我们来看 如何将探针名称转换为基因名称。






![[Machine learning][Part3] numpy 矢量矩阵操作的基础知识](https://img-blog.csdnimg.cn/5ebe08b97ac94f8283df2adfd4a95054.png)
