第六节,我们使用结核病基因数据,做了一个数据预处理的实操案例。例子中结核类型,包括结核,潜隐进展,对照和潜隐,四个类别。第七节延续上个数据,进行了差异分析。 本节对差异基因进行富集分析。
目录
数据展示
GO富集分析 -对基因名称映射基因ID
GO富集分析 -从org.Hs.eg.db库中去匹配基因
KEGG富集分析 (不详细讲了看注释)
GSEA 富集分析
更多复杂的图(关联网络图、八卦图 、弦图)
数据展示
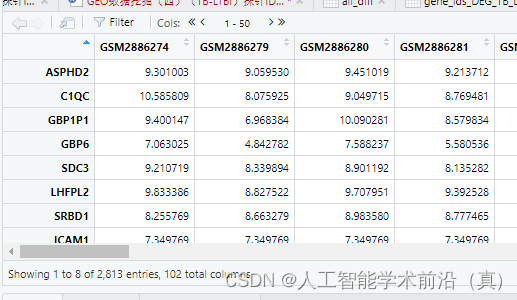
差异基因计算完毕的指标如下图所示

差异基因筛选后表达矩阵
GO富集分析 -对基因名称映射基因ID
加载数据
#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#+&&&&&&&&&&&&&&&&&&加载数据&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
load( "DEG_TB_LTBI_step13.Rdata")
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
#+&&&&&&&&&&&&&&&&&&加载数据&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&library(clusterProfiler)
library(org.Hs.eg.db)#增加基因名
all_diff$SYMBOL=rownames(all_diff)#基因名称转换注释
gene_ids_DEG_TB_LTBI = bitr(geneID = rownames(dataset_TB_LTBI_DEG),fromType="SYMBOL",toType = c("ENTREZID","ENSEMBL","SYMBOL"),OrgDb = 'org.Hs.eg.db',drop =TRUE)#合并 增加logFC 为后续GSEA富集分析所需数据准备
gene_ids_DEG_TB_LTBI <- merge(gene_ids_DEG_TB_LTBI,all_diff,by="SYMBOL")#观察
dim(gene_ids_DEG_TB_LTBI)
head(gene_ids_DEG_TB_LTBI)#获取基因ID ENSEMBL
gene_ENSEMBL <- gene_ids_DEG_TB_LTBI$ENSEMBL
gene_ENTREZID <- gene_ids_DEG_TB_LTBI$ENTREZID
gene_SYMBOL<- gene_ids_DEG_TB_LTBI$SYMBOL
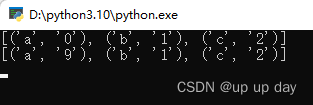
经过映射,2813个差异基因得到2551个基因ID,下图为三种不同形式的基因名称,富集分析时,按需进行转换。

GO富集分析 -从org.Hs.eg.db库中去匹配基因
#Go富集分析,从库中去匹配
go <- enrichGO(gene_SYMBOL,OrgDb = org.Hs.eg.db, ont='ALL',pAdjustMethod = 'BH',pvalueCutoff = 0.05, qvalueCutoff = 0.2,keyType = 'SYMBOL')#进行GO富集,确定P值与Q值得卡值并使用BH方法对值进行调整。
#查看富集结果
dim(go)
#导出GO富集的结果
write.csv(go,file="go1.csv")绘制气泡图
#绘制气泡图
pdf(file="15aGO富集分析step15.pdf", width = 9, height = 6)
dotplot(go,showCategory=20,label_format = 80)#气泡图dev.off()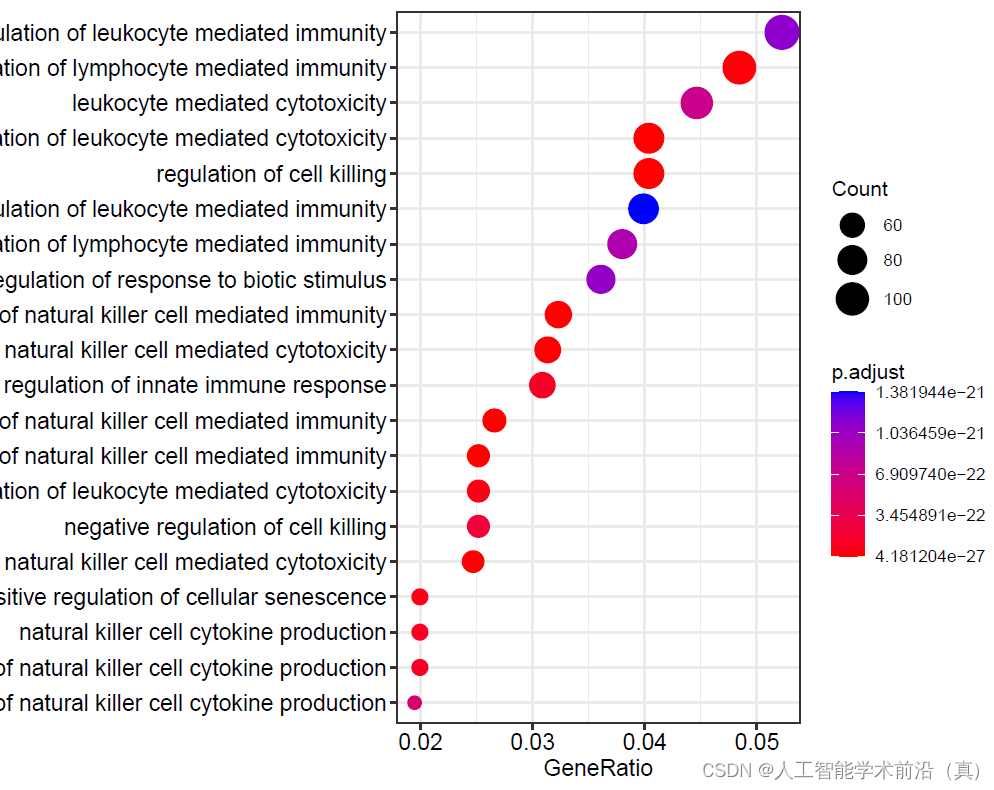
三种不同类别的合并的气泡图(#CC细胞组件,MF分子功能,BP生物学过程)
pdf(file="15bGO富集分析三组step15.pdf", width = 9, height = 6)#CC细胞组件,MF分子功能,BP生物学过程
goCC <- enrichGO(gene = gene_ENTREZID, #基因列表(转换的ID)keyType = "ENTREZID", #指定的基因ID类型,默认为ENTREZIDOrgDb=org.Hs.eg.db, #物种对应的org包ont = "CC", #CC细胞组件,MF分子功能,BP生物学过程pvalueCutoff = 0.05, #p值阈值pAdjustMethod = "fdr", #多重假设检验校正方式minGSSize = 1, #注释的最小基因集,默认为10maxGSSize = 500, #注释的最大基因集,默认为500qvalueCutoff = 0.2, #q值阈值readable = TRUE) #基因ID转换为基因名goBP <- enrichGO(gene_ENTREZID,OrgDb = org.Hs.eg.db, ont='BP',pAdjustMethod = 'BH',pvalueCutoff = 0.05, qvalueCutoff = 0.2,keyType = 'ENTREZID')goMF <- enrichGO(gene_ENTREZID,OrgDb = org.Hs.eg.db, ont='MF',pAdjustMethod = 'BH',pvalueCutoff = 0.05, qvalueCutoff = 0.2,keyType = 'ENTREZID')
#通过ggplot2将BP、MF、CC途径的富集结果挑选前8条绘制在一张图上
barplot(go,label_format=100, split="ONTOLOGY")+ facet_grid(ONTOLOGY~.,scale="free")dev.off()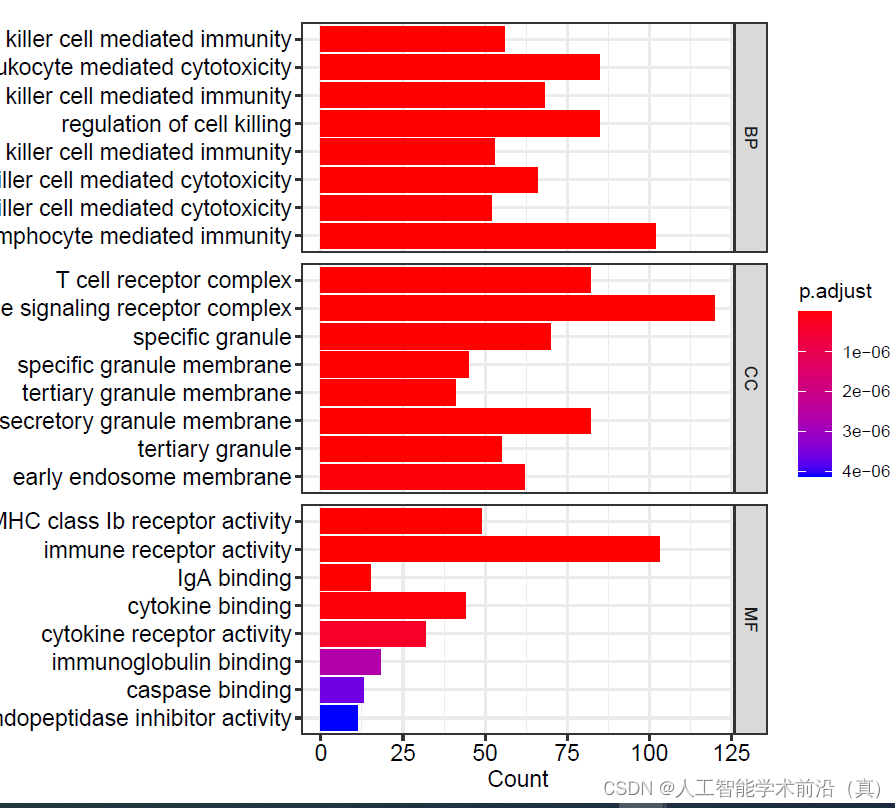
KEGG富集分析 (不详细讲了看注释)
#+=================================================================
#============================================================
#+========KEGG富集分析 气泡图step16===================
#+==========================================
#+================================#KEGG富集分析
pdf(file="16KEGG富集分析step16.pdf", width = 9, height = 6)
kegg<- enrichKEGG(gene = gene_ENTREZID, #基因列表(ENTREZID ID: 54490,51144,31,3906) organism = "hsa", #物种keyType = "kegg", #指定的基因ID类型,默认为keggminGSSize = 3, maxGSSize = 500,pvalueCutoff = 0.05, pAdjustMethod = "fdr", # pAdjustMethod = 'BH'qvalueCutoff = 0.02)
#观察
dim(kegg)
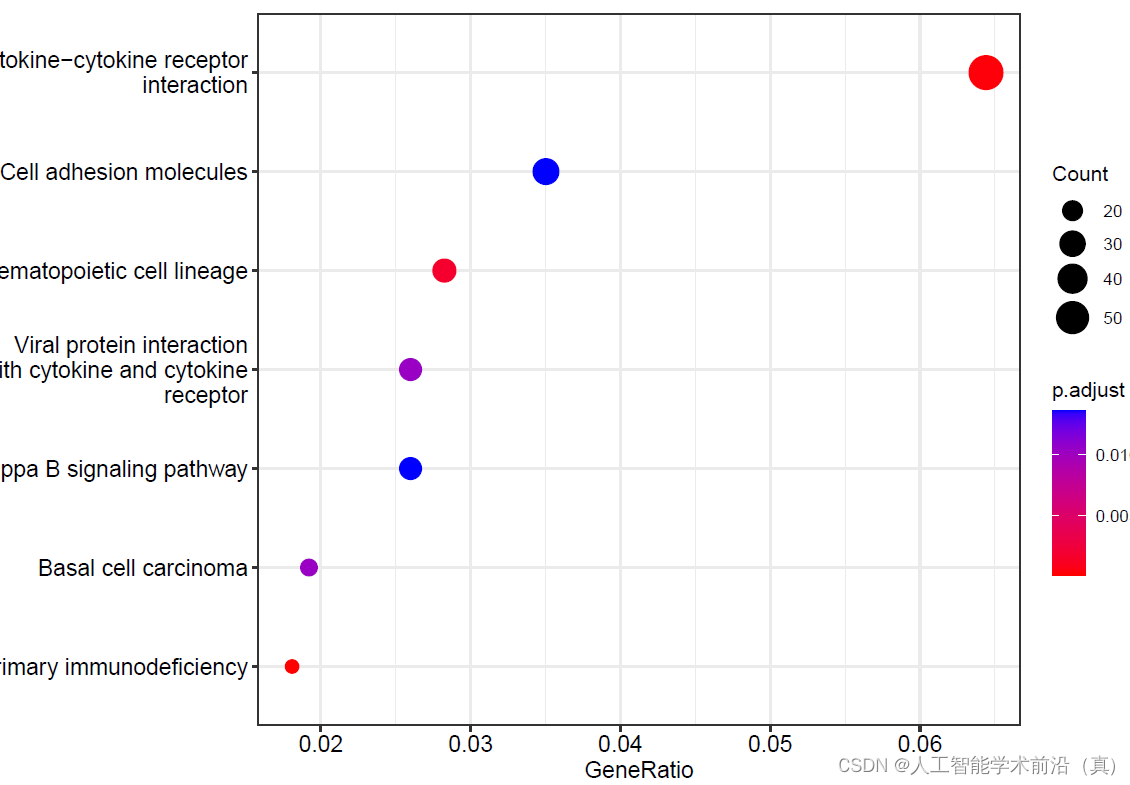
#绘制气泡图
dotplot(kegg)
dev.off()#kegg 增加可读性,对基因ID 转基因名
kegg_enrich_results <- DOSE::setReadable(kegg, OrgDb="org.Hs.eg.db", keyType='ENTREZID') #ENTREZID to gene Symbol#保存kegg结果
write.csv(kegg_enrich_results@result,'KEGG_gene_up_enrichresults.csv')
#save(kegg_enrich_results, file ='KEGG_gene_up_enrichresults.Rdata')##查看与选择所需通路
kegg_enrich_results@result$Description[1:10] #查看前10通路###选择所需通路的ID号
i=1
select_pathway <- kegg_enrich_results@result$ID[i] #选择所需通路的ID号#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#+&&&&&&&&&&&&&&&&&&数据保存&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
save(gene_ids_DEG_TB_LTBI,go,keggfile ="15_gene_ids_DEG_TB_LTBI.Rdata")
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
#+&&&&&&&&&&&&&&&&&&数据保存&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
GSEA 富集分析
#+=================================================================
#============================================================
#+========GSEA 富集分析 气泡图step17===================
#+==========================================
#+================================# GSEA 分析
#需要把多个方法取并集
#该方法的输入需要基因和 logFC 排序后的结果
#不同方法 相同基因的的logFC值不一样,直接保留第一个重复基因library(stringr)## 去重 #去除NA值
dim(gene_ids_DEG_TB_LTBI)
colnames(gene_ids_DEG_TB_LTBI)gene_list_df = gene_ids_DEG_TB_LTBI[,c('ENTREZID','logFC')]
gene_list_df_na <- na.omit(gene_list_df)gene_ids_TB_LTBI_distinct <- dplyr::distinct(gene_list_df_na,ENTREZID,.keep_all=TRUE) dim(gene_ids_TB_LTBI_distinct)
gene_list=gene_ids_TB_LTBI_distinct$logFC #提取logFC列
names(gene_list)=gene_ids_TB_LTBI_distinct$ENTREZID #加上ENTREZID
gene_list_gsea = sort(gene_list, decreasing = T) #降序排列gsea_KEGG <- gseKEGG(gene_list_gsea,organism = "hsa",keyType = "kegg")gsea_KEGG_d <- as.data.frame(gsea_KEGG)gsea_KEGG_d
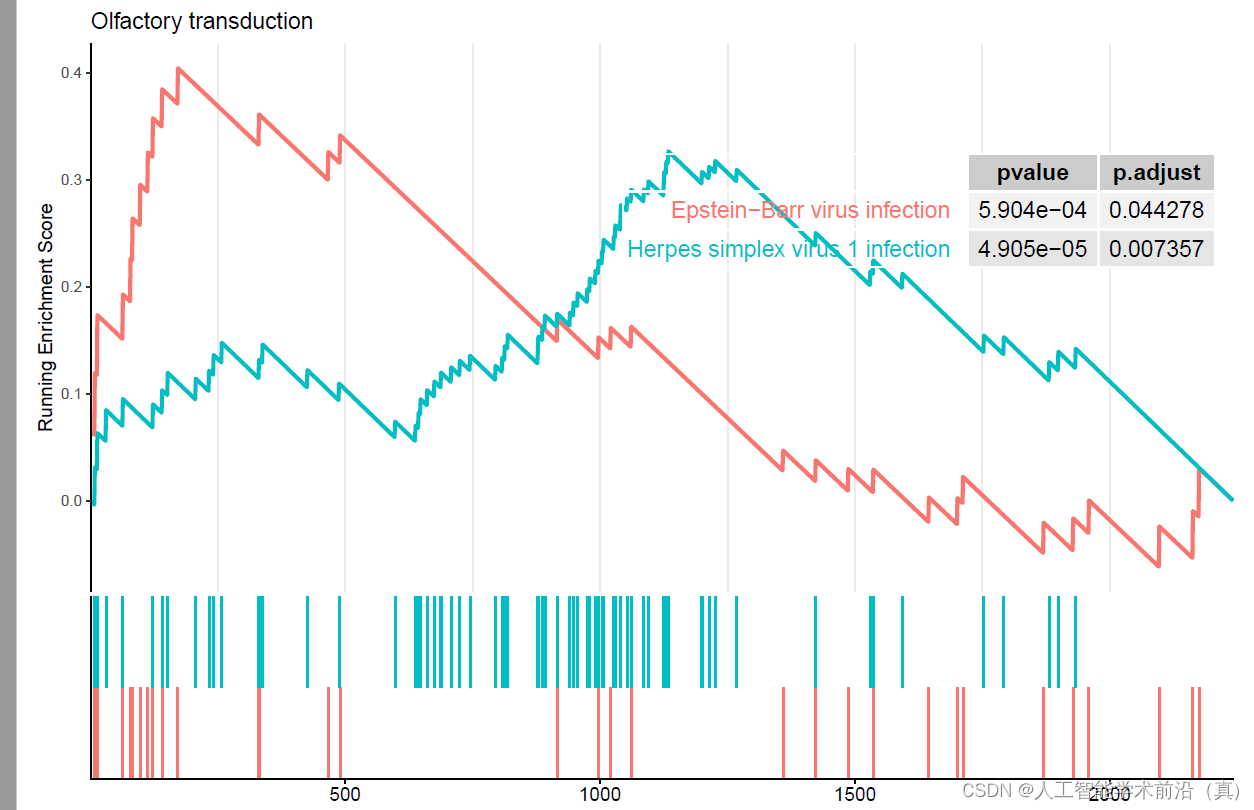
#path 为需要展示的pathway id,这里展示的是enrichment score最高的4条通路t_index=order(gsea_KEGG_d$enrichmentScore,decreasing = T)
path=rownames(gsea_KEGG[t_index,]) #选择展示的 pathwayrownames(gsea_KEGG[t_index,]) [1:4]#作图
pdf(file="17GSEA富集分析step17.pdf", width = 9, height = 6)
gseaplot2(gsea_KEGG,path, subplots = 1:2, #展示前2个图pvalue_table = T, #显示p值title = "Olfactory transduction", #设置titlebase_size = 10, #字体大小color="red") #线条颜色可选
dev.off()#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#+&&&&&&&&&&&&&&&&&&数据保存&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
save(gene_ids_DEG_TB_LTBI,go,kegg,gene_list_gsea,gsea_KEGG,file ="17_gene_ids_DEG_TB_LTBI.Rdata")
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
#+&&&&&&&&&&&&&&&&&&数据保存&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
更多复杂的图(关联网络图、八卦图 、弦图)
参考 最全的GO, KEGG, GSEA分析教程(R),你要的高端可视化都在这啦![包含富集圈图] - 糖糖家的老张的文章 - 知乎 https://zhuanlan.zhihu.com/p/377356510
原文链接:https://blog.csdn.net/qq_50898257/article/details/120588222
#+=================================================================
#============================================================
#+========富集分析 更多的图step18===================
#+==========================================
#+================================
library(clusterProfiler)
library(enrichplot)
#+富集基因与所在功能集/通路集的关联网络图:
enrichplot::cnetplot(go,circular=FALSE,colorEdge = TRUE)#基因-通路关联网络图
enrichplot::cnetplot(kegg,circular=FALSE,colorEdge = TRUE)#circluar为指定是否环化,基因过多时建议设置为FALSEGO2 <- pairwise_termsim(go)
KEGG2 <- pairwise_termsim(kegg)
enrichplot::emapplot(GO2,showCategory = 50, color = "p.adjust", layout = "kk")#通路间关联网络图
enrichplot::emapplot(KEGG2,showCategory =50, color = "p.adjust", layout = "kk")write.table(kegg$ID, file = "KEGG_IDs.txt", #将所有KEGG富集到的通路写入本地文件查看append = FALSE, quote = TRUE, sep = " ",eol = "\n", na = "NA", dec = ".", row.names = TRUE,col.names = TRUE, qmethod = c("escape", "double"),fileEncoding = "")
#打印几条通路名称看看
kegg$ID[1:3]
#打开浏览器观察通路
browseKEGG(kegg,"hsa04660")#选择其中的hsa05166通路进行展示#富集弦图
genedata<-data.frame(ID=gene_ids_DEG_TB_LTBI$SYMBOL ,logFC=gene_ids_DEG_TB_LTBI$logFC)
write.table(go$ONTOLOGY, file = "GO_ONTOLOGYs.txt", #将所有GO富集到的基因集所对应的类型写入本地文件从而得到BP/CC/MF各自的起始位置如我的数据里是1,2103,2410append = FALSE, quote = TRUE, sep = " ",eol = "\n", na = "NA", dec = ".", row.names = TRUE,col.names = TRUE, qmethod = c("escape", "double"),fileEncoding = "")'''
根据计算出的go 文件数量,调整
'''
GOplotIn_BP<-go[1:178,c(2,3,7,9)] #提取GO富集BP的前10行,提取ID,Description,p.adjust,GeneID四列
GOplotIn_CC<-go[179:194,c(2,3,7,9)]#提取GO富集CC的前10行,提取ID,Description,p.adjust,GeneID四列
GOplotIn_MF<-go[195:209,c(2,3,7,9)]#提取GO富集MF的前10行,提取ID,Description,p.adjust,GeneID四列library(stringr)
GOplotIn_BP$geneID <-str_replace_all(GOplotIn_BP$geneID,'/',',') #把GeneID列中的’/’替换成‘,’
GOplotIn_CC$geneID <-str_replace_all(GOplotIn_CC$geneID,'/',',')
GOplotIn_MF$geneID <-str_replace_all(GOplotIn_MF$geneID,'/',',')
names(GOplotIn_BP)<-c('ID','Term','adj_pval','Genes')#修改列名,后面弦图绘制的时候需要这样的格式
names(GOplotIn_CC)<-c('ID','Term','adj_pval','Genes')
names(GOplotIn_MF)<-c('ID','Term','adj_pval','Genes')
GOplotIn_BP$Category = "BP"#分类信息
GOplotIn_CC$Category = "CC"
GOplotIn_MF$Category = "MF"BiocManager::install('GOplot')
library(GOplot)
circ_BP<-GOplot::circle_dat(GOplotIn_BP,genedata) #GOplot导入数据格式整理
circ_CC<-GOplot::circle_dat(GOplotIn_CC,genedata)
circ_MF<-GOplot::circle_dat(GOplotIn_MF,genedata) chord_BP<-chord_dat(data = circ_BP,genes = genedata) #生成含有选定基因的数据框
chord_CC<-chord_dat(data = circ_CC,genes = genedata)
chord_MF<-chord_dat(data = circ_MF,genes = genedata)
'''
> chord_CC<-chord_dat(data = circ_CC,genes = genedata)
Error in `[<-`(`*tmp*`, g, p, value = ifelse(M[g] %in% sub2$genes, 1, : subscript out of bounds我去检查了go和genelist的数据结构发现,genelist里的gene用的是gene名,而go里的基因用的是基因ID,不一样了,所以跑不出结果,所以我把genelist的gene换成了基因ID,就能跑出来了。作者:ff的小世界勿扰
链接:https://www.jianshu.com/p/ee4012fd253f
来源:简书
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。
'''#可以画 数量太多了
GOChord(data = chord_BP,#弦图title = 'GO-Biological Process',space = 0.01,#GO Term间距limit = c(1,1),gene.order = 'logFC',gene.space = 0.25,gene.size = 5,lfc.col = c('red','white','blue'), #上下调基因颜色process.label = 10) #GO Term字体大小
GOChord(data = chord_CC,title = 'GO-Cellular Component',space = 0.01,limit = c(1,1),gene.order = 'logFC',gene.space = 0.25,gene.size = 5,lfc.col = c('red','white','blue'), process.label = 10)
GOChord(data = chord_MF,title = 'GO-Mollecular Function',space = 0.01,limit = c(1,1),gene.order = 'logFC',gene.space = 0.25,gene.size = 5,lfc.col = c('red','white','blue'), process.label = 10)
'''
Warning messages:
1: Using size for a discrete variable is not advised.
2: Removed 15 rows containing missing values (`geom_point()`).
'''富集分析完毕!
回顾我们用到方法,差异分析后进行富集分析,理论基础实际上就是简单的找不同,分析。
实际应用种,由于基因之间存在关联,另一套分析理论考虑的是基因之间的相互作用,下节,我们来看非常火的WGCNA 共表达加权网络进行基因分析。