❝写在前面
由于最近在提交课题数据到 NCBI 数据库,整理了相关笔记。本着自己学习、分享他人的态度,分享学习笔记,希望能对大家有所帮助。推荐先按顺序阅读往期内容:
1. 提交高通量测序数据到 GEO --- 说明书
2. 提交高通量测序原始数据到 SRA --- 操作流程
目录
-
1 注册 NCBI 账号 -
2 准备要上传的数据 -
2.1 Raw data files -
2.2 Processed data files -
2.3 Metadata spreadsheet
-
-
3 上传数据
在发表文章之前往往需要将高通量测序的数据上传到 NCBI 数据库。上传的数据可以分为两类:① Raw data(fastq文件)通常上传到 SRA、② Processed data(counts matrix、RDS 等)通常上传到 GEO。本文详细介绍提交高通量测序 Processed data 到 GEO 的操作流程。
在进行操作之前,强烈建议先阅读我之前的推文:《提交高通量测序数据到 GEO --- 说明书》。里面详细介绍了要准备的文件和格式要求。
1 注册 NCBI 账号
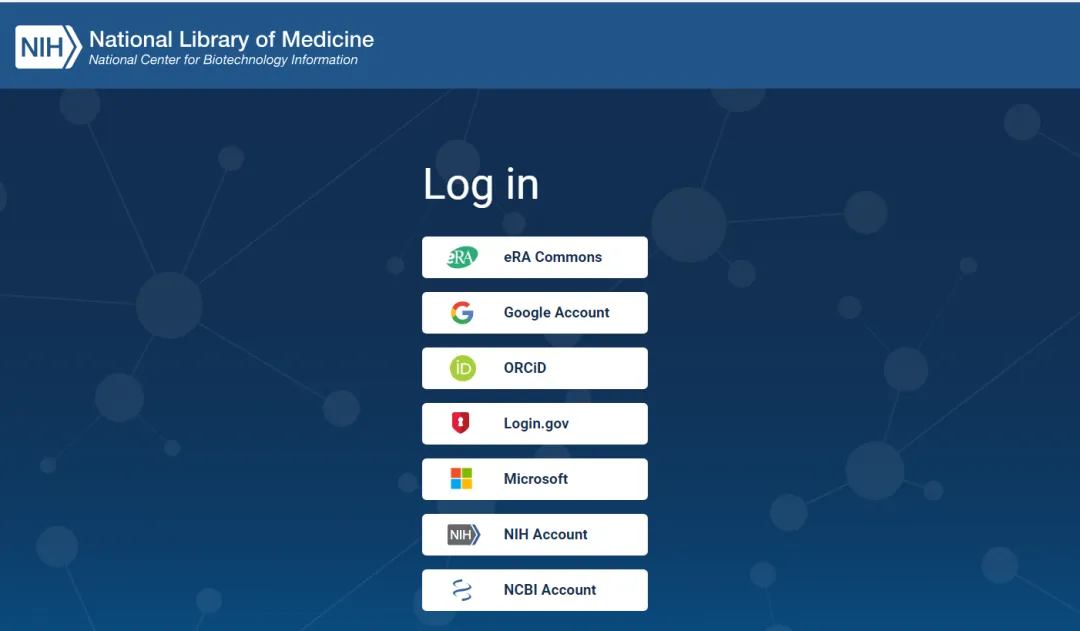
首先需要登陆https://www.ncbi.nlm.nih.gov/account/注册一个 NCBI 账号,NCBI 支持用各种第三方账户进行注册:
2 准备要上传的数据
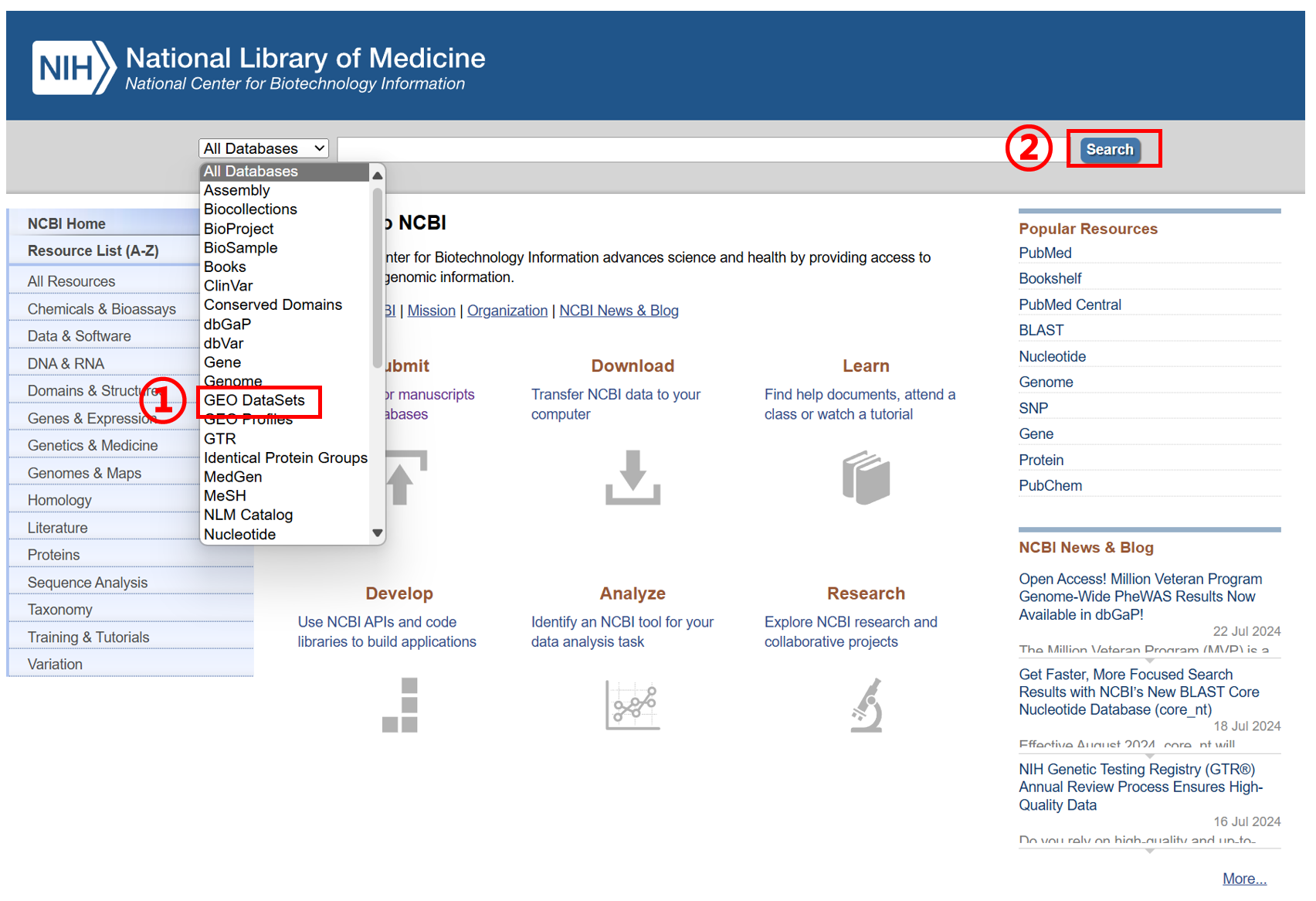
进入 NCBI 首页(https://www.ncbi.nlm.nih.gov/),按如下操作:① 选择 GEO DataSets、② 点击 Search、③ 点击 Submit to GEO、④ Submit high-throughput sequencing
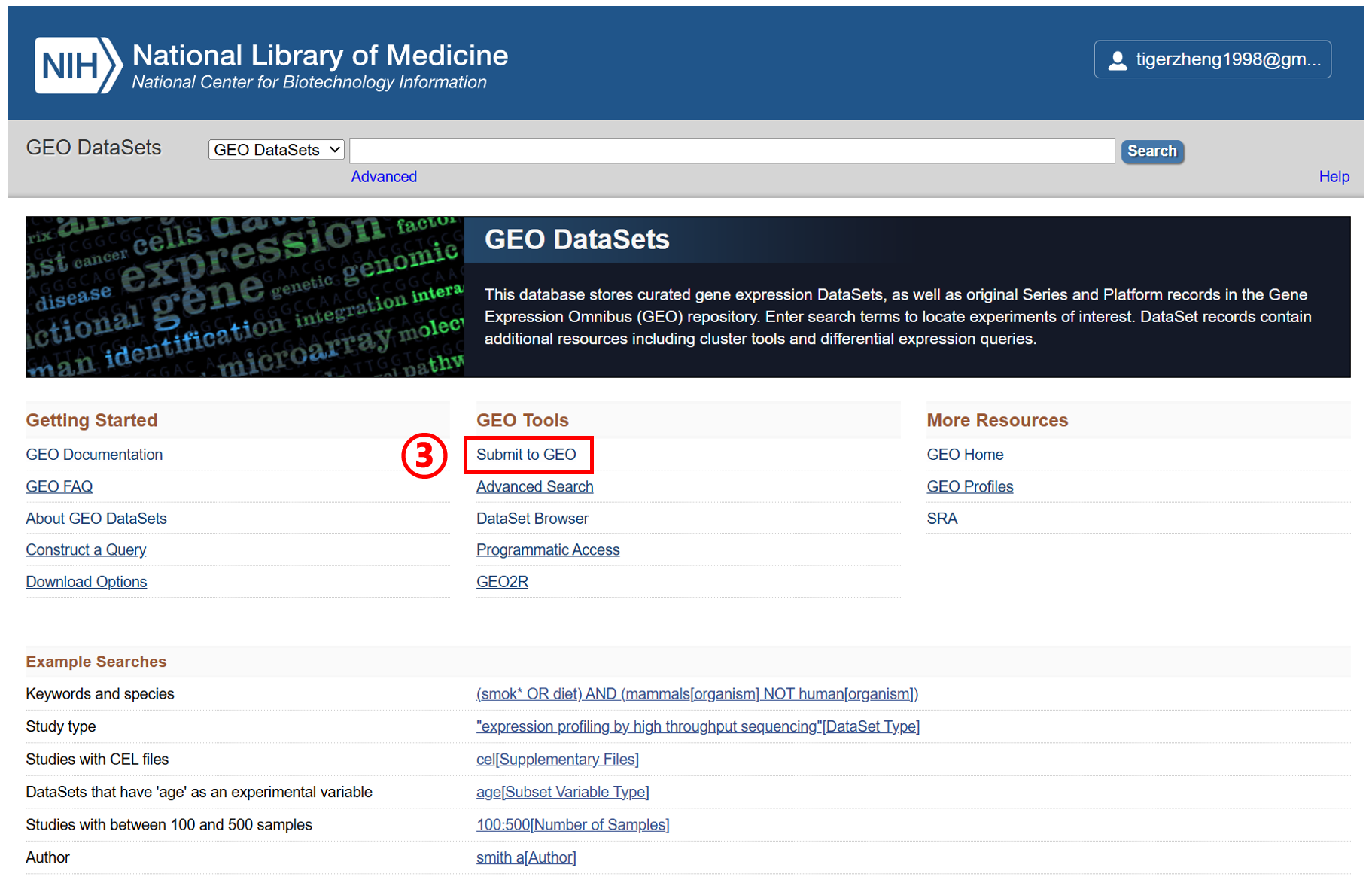
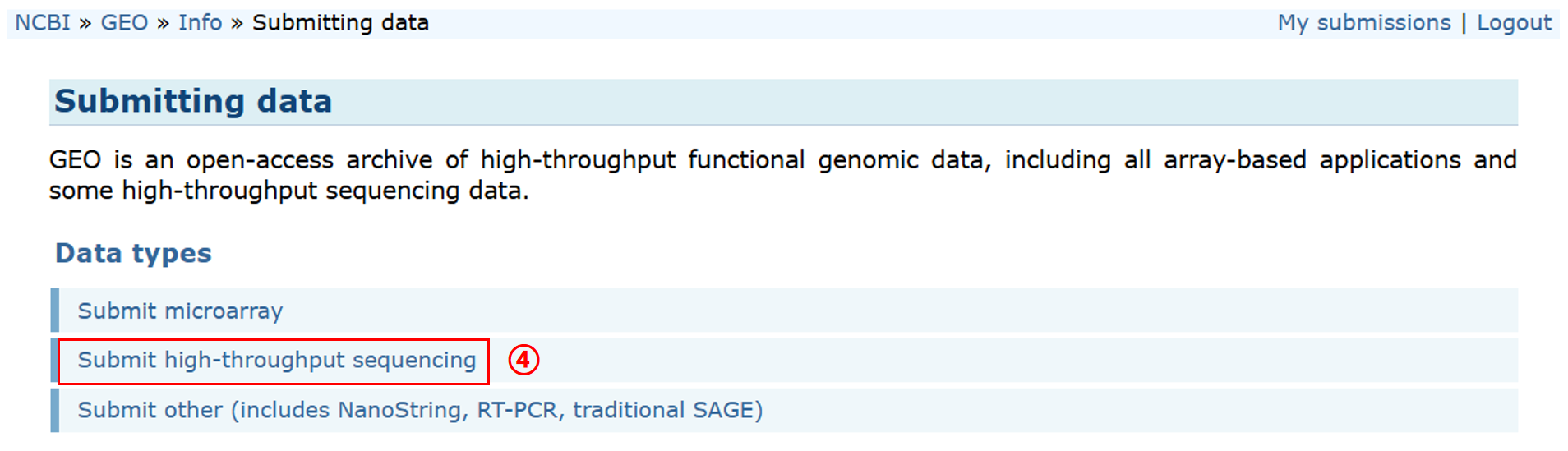
可以看到需要准备以下数据:1. Metadata spreadsheet、2. Processed data files、3. Raw data files

2.1 Raw data files
Raw data 是测序数据的 fastq 文件,可以将 raw data 提交给 GEO,也可以直接提交给 SRA。但是 raw data 往往非常大,按照 GEO 推荐的 FileZilla 软件上传非常的慢,因此推荐用 Aspera 命令行直接将 raw data 上传到 SRA。Aspera 提供跨越洲际距离的快速上传连接,上传速度可达 100Mb/s。
关于如何将 raw data 上传到 SRA,可以查看我之前的推文:《提交高通量测序原始数据到 SRA --- 操作流程》
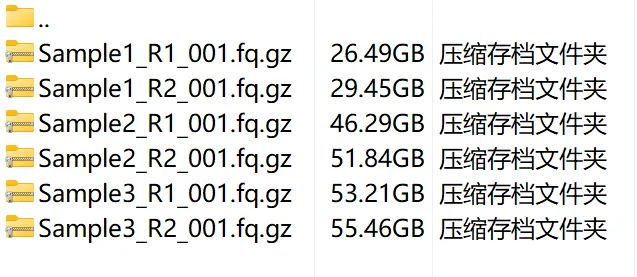
2.2 Processed data files
Processed data 一般为比对后生成的 count matrix 文件。比如我这里为单细胞测序数据比对后的 cell-gene 矩阵的 csv 文件:

2.3 Metadata spreadsheet
Metadata 是指有关研究、样本、方法以及对 processed data 和 raw data 文件名的引用的描述性信息。GEO 中提供了两种模板,由于我这里选择将 raw data 提交到 SRA,因此选择 "Download metadata spreadsheet with SRA accessions"。

2.3.1 STUDY
STUDY 部分介绍研究的整体概况。STUDY 部分提供的信息将在公共网页上的 GEO Series(GSE record)中显示。

-
title:标题,可以是发表文章的标题。 -
summary(abstract):摘要,可以是发表文章的摘要。 -
experimental design:实验设计,描述 submission 中包括的样本类型,已经实验条件和变量。包括分析什么类型样本、是否包括复制、是否有对照和/或参考样本等。不包括 protocols/methods。可以参照 EXAMPLE sheets 填写。 -
contributor:作者,例如 Yixin, Wang,每个作者都在单独的一行上。 -
supplementary file:补充文件,如果您提交的 processed data 文件对应于多个样本。例如:fpkms_allsamples.txt。该文件应该具有与 metadata SAMPLES 中的唯一描述符匹配的唯一列名(例如,"library name")。single-cell submissions 除外。 -
BioProject:BioProject accession,例如:PRJNA123456
2.3.2 SAMPLES
SAMPLES 部分是对样本的描述。SAMPLES 部分提供的信息将在公共网页上的 GEO Samples(GSM records)中显示。
注意:*为必填项,**为至少选择一项填写


-
library name:文库名称,必须唯一,如 Sample1、Sample2... -
title:样本名称,必须唯一,建议使用以下约定: [biomaterial][condition(s)][replicate number],例如:Muscle, exercised,60min, rep2 -
organism:物种,如 Mus musculus -
BioSample:BioSample accessions,如 SAMN123456 -
SRA Experiment or Run:SRX/SRR accessions,如 SRR123456 -
tissue:组织,如 Distal colon -
cell line:细胞系,如 C3H 10T1/2 -
cell type:细胞类型,如 mesenchymal stem cells -
genotype:基因型,如 WT、KDM4D knockdown -
treatment:处理,如 adipogenic differentiation -
molecule:分子类型,选择以下之一:total RNA、polyA RNA、cytoplasmic RNA、nuclear RNA、genomic DNA、protein、other -
single or paired-end:测序类型,选择 single 或 paired-end -
instrument model:仪器型号,如 Illumina NovaSeq 6000。 -
description:描述信息,可选的,其他选项中没有提供的额外信息。或者,如果您提交的矩阵包含多个样品的 processed data (例如,所有 RNA-Seq 样品的 Counts.txt),请在此处列出矩阵列名称。 -
processed data file:包含 processed data 的文件的确切名称。例如:HeLa_H3K4me3.bw。可以为多个样本列出相同的处理文件。如果一个样本存在多个处理文件,则可以包括多个 'processed data file' 列。
2.3.3 PROTOCOLS
PROTOCOLS 部分是对实验方法的描述。PROTOCOLS 部分提供的信息将出现在每个 GEO Sample(GSM record)中。

-
growth protocol:可选项,描述在提取前用于培养或维持生物体或细胞的条件。 -
treatment protocol:可选项,描述在提取制备之前对生物材料进行的处理。 -
extract protocol:必填项,描述用于提取和准备待测序材料的方法。 -
library construction protocol:必填项,描述文库构建方法。 -
library strategy:必填项,描述每个文库的测序技术,如 RNA-Seq。 -
data processing step:必填项,提供有关如何生成 processed data 文件的详细信息。步骤可能包括:基本调用软件、版本、参数;数据过滤步骤;Read 比对软件版本、参数;额外的处理软件(例如,peak-calling, abundancemeasurement)、版本、参数;等。 -
genome build/assembly:必填项,UCSC或NCBI基因组构建号(例如,hg18、mm9、human NCBI genome build 36 ...),或用于 read 比对的参考序列。 -
processed data files format and content:必填项,对于每种 processed data 文件类型,提供格式和内容的描述。如 Tab-separated values files and matrix files。
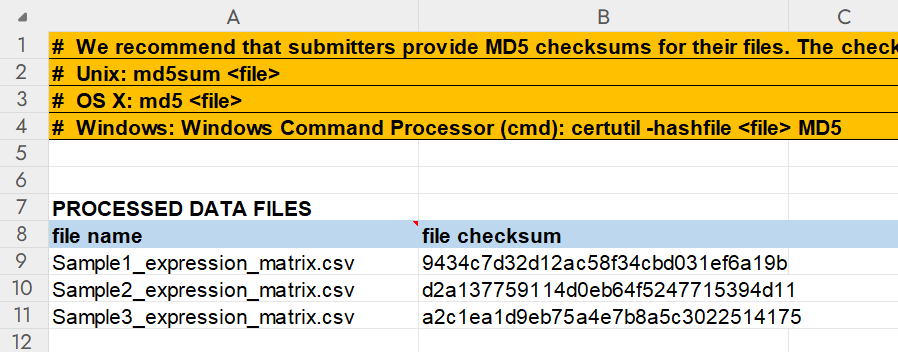
2.3.4 MD5 Checksums
在填写完 Metadata 页面后,还需要在 MD5 Checksums 页面填写所有 processed data 文件的 MD5 校验码。checksums 可以使用以下方法计算:
-
Unix: md5sum <file> -
OS X: md5 <file> -
Windows: Windows Command Processor (cmd): certutil -hashfile <file> MD5
这里以 Windows 上命令行为例:
打开 cmd,输入以下命令:certutil -hashfile "F:\path\Sample1_expression_matrix.csv" MD5
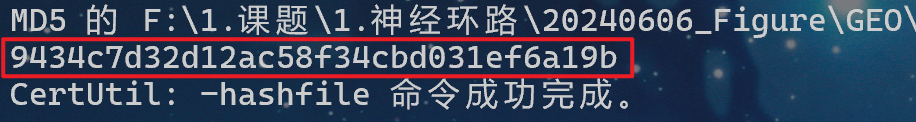
将文件名和生成的MD5码填入表格:
3 上传数据
点击 Transfer files 开始上传数据。
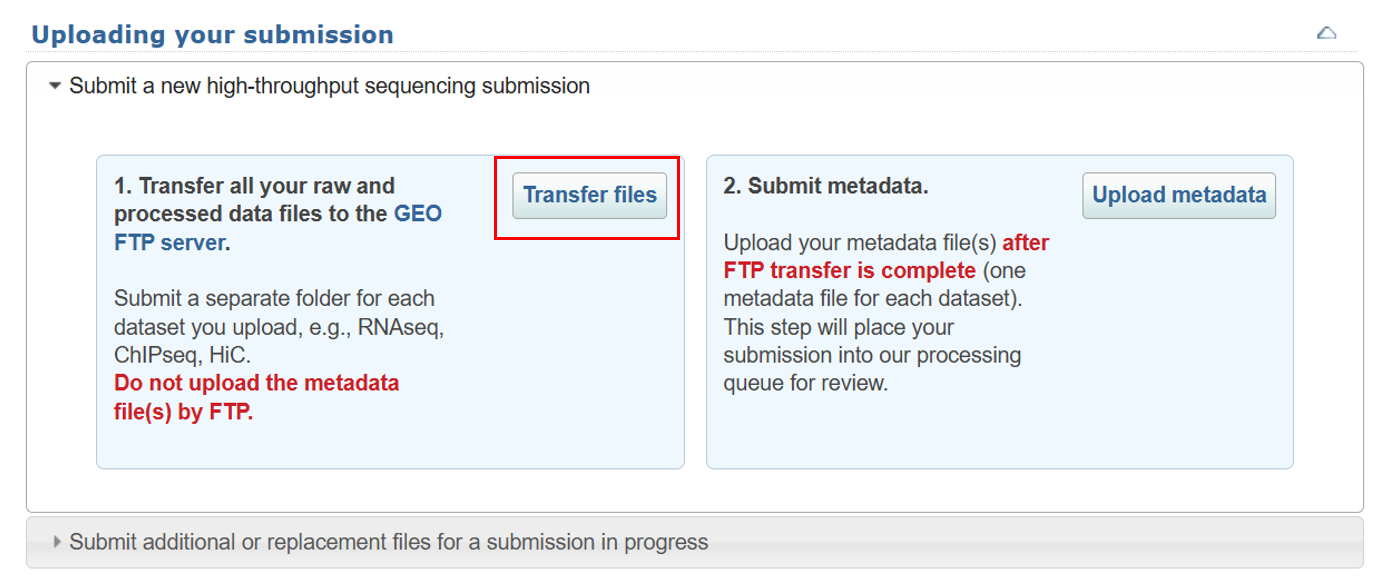
进入后可以看到上传数据的个人空间地址,以及 FTP 服务器账号和密码。
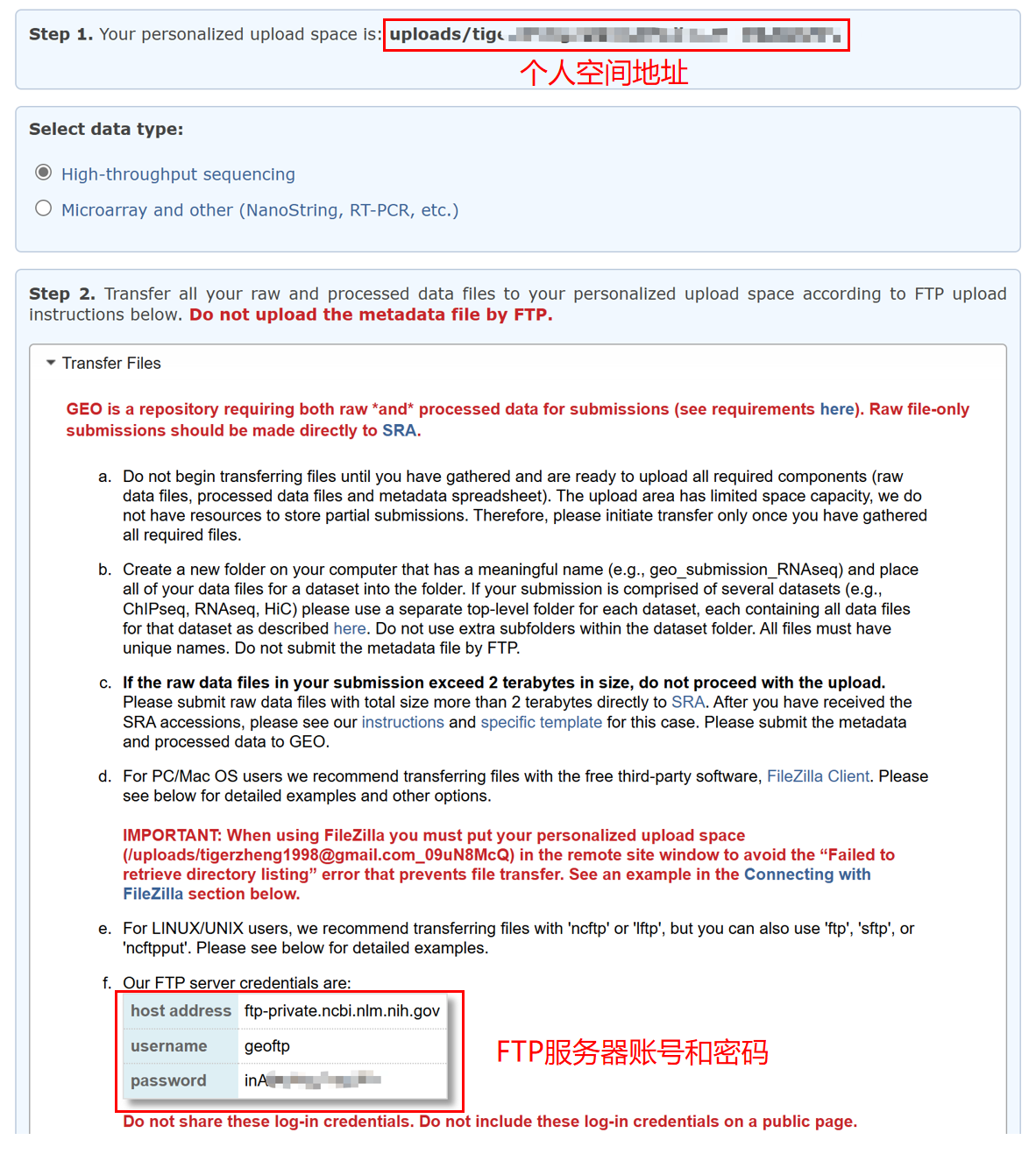
下载 GEO 推荐的第三方文件传输软件 FileZila Client,下载地址:https://filezilla-project.org/download.php?show_all=1。
下载后按下图操作:① 输入主机地址、用户名、密码;② 点击快速连接;③ 输入个人空间地址;④ 找到需上传文件地址;⑤ 将需上传文件拖拽到此处
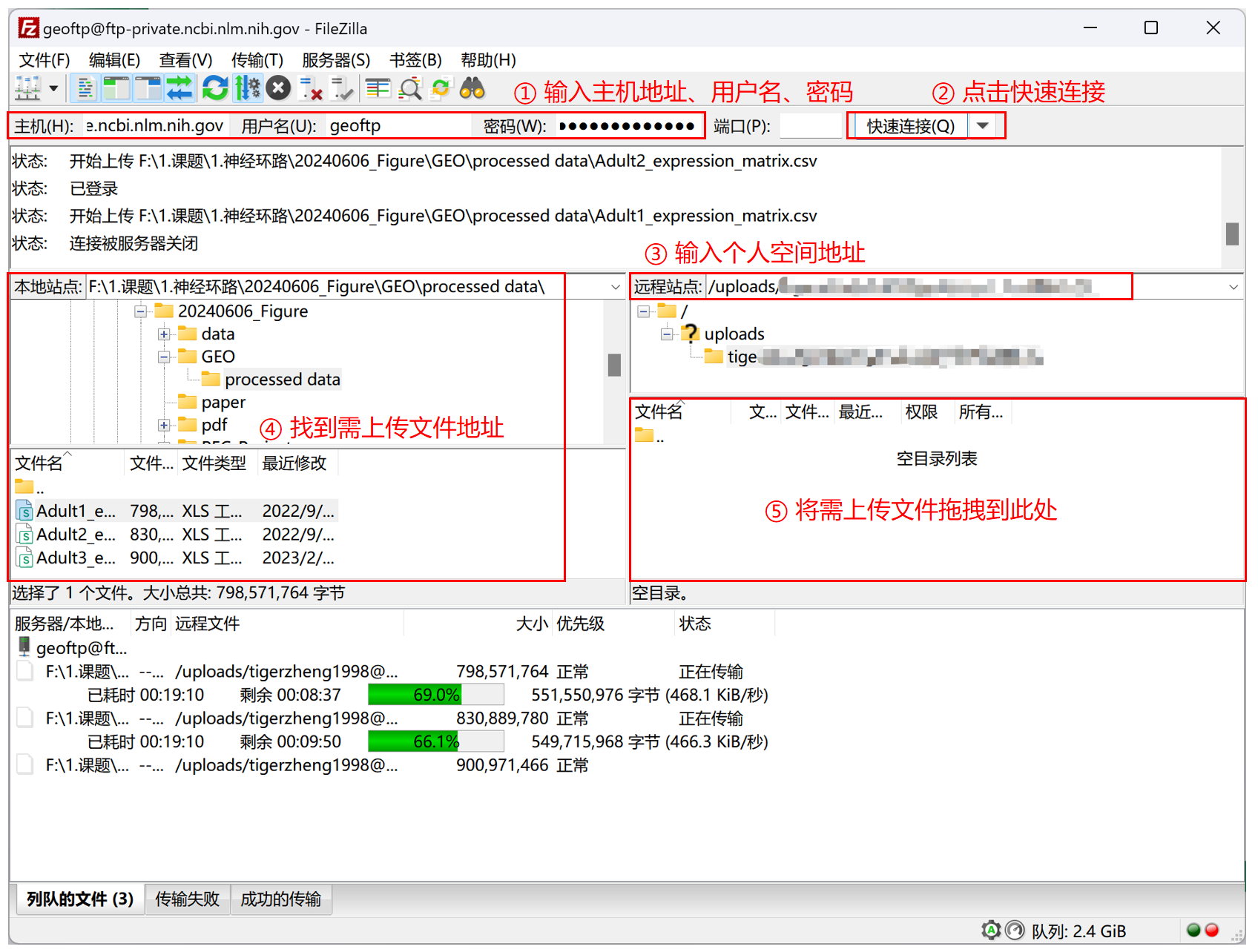
可以看到上传速度非常慢(0.5Mb/s),需要等待很长时间。这也是为什么不推荐将原始 fastq 文件上传到 GEO。荐用 Aspera 命令行直接将 fastq 文件上传到 SRA,上传速度可达 100Mb/s。
数据上传完成之后,上传 metadata 文件:
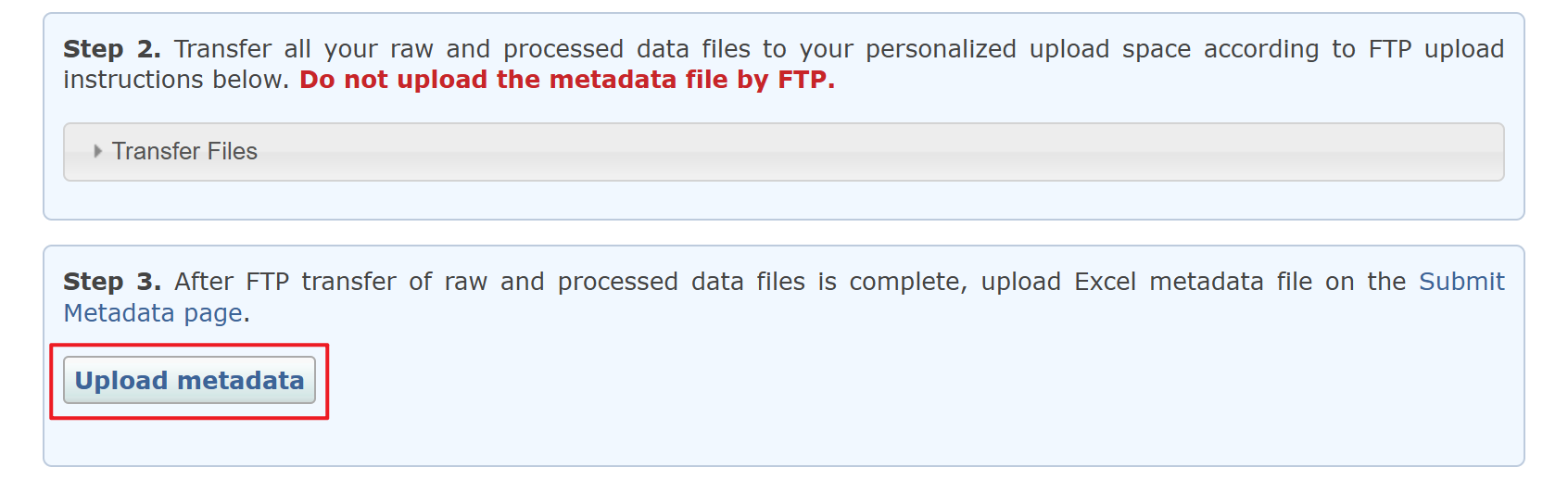
选择前面填写的 metadata Excel 文件,选择发布日期,然后点 Submit 提交。
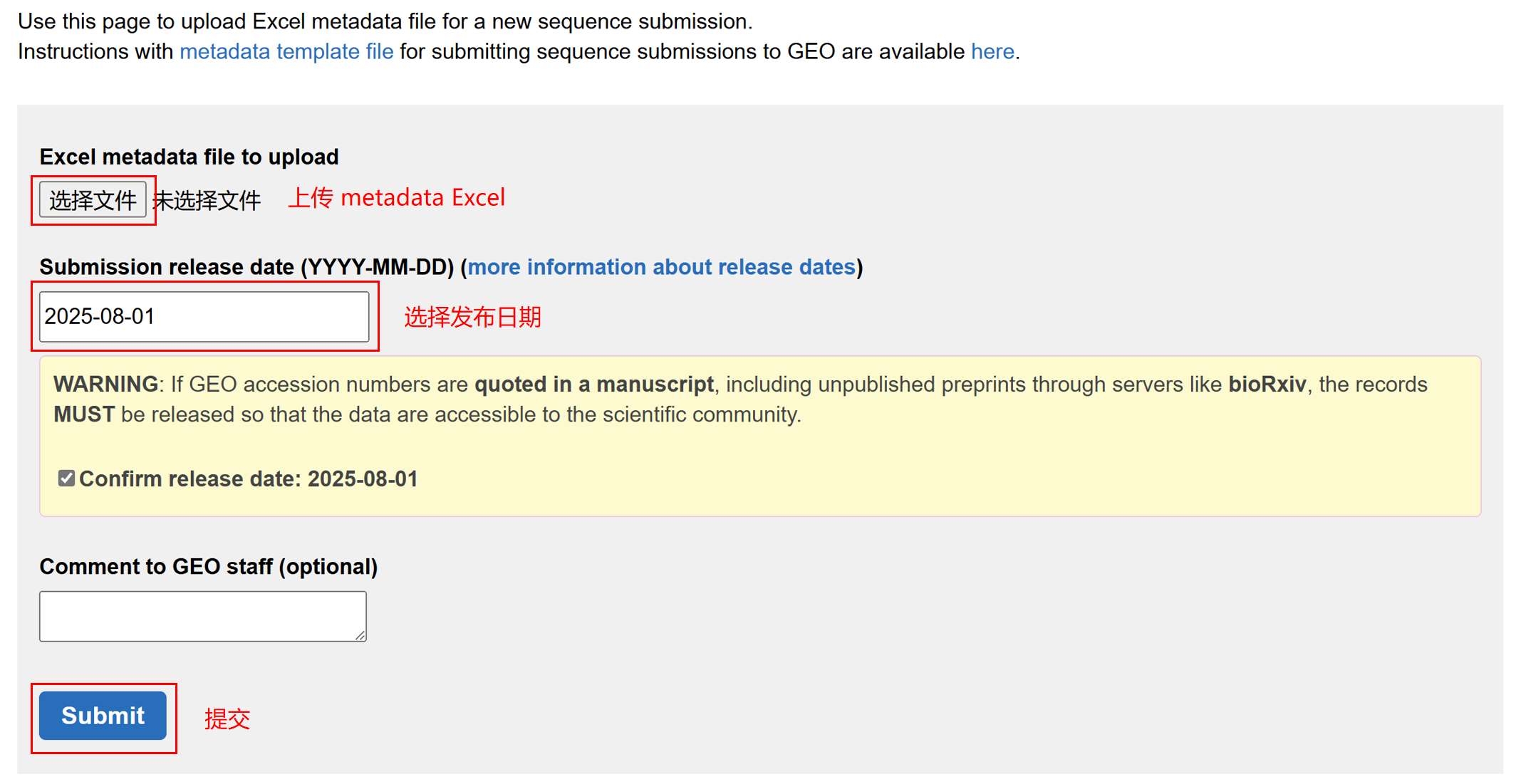
一般提交后一天内就会收到邮件,看到 GEO 编号,状态为 resolved,就表示数据提交成功了。邮件中还提供了后续如何更改信息,以及更改发布日期的相关连接。

注:本文为个人学习笔记,仅供大家参考学习,不得用于任何商业目的。如有侵权,请联系作者删除。

本文由 mdnice 多平台发布


![[FBCTF2019]RCEService (PCRE回溯绕过和%a0换行绕过)](https://i-blog.csdnimg.cn/direct/ed5f04508d4041af82c9770d26abed10.png)