点击蓝字 关注我们
使用PhyloSuite进行分子系统发育及系统发育树的统计分析

iMeta主页:http://www.imeta.science
方 法
● 原文链接DOI: https://doi.org/10.1002/imt2.87
● 2023年2月16日,兰州大学张东团队在 iMeta 在线发表了题为“Using PhyloSuite for molecular phylogeny and tree-based analyses ”的文章。
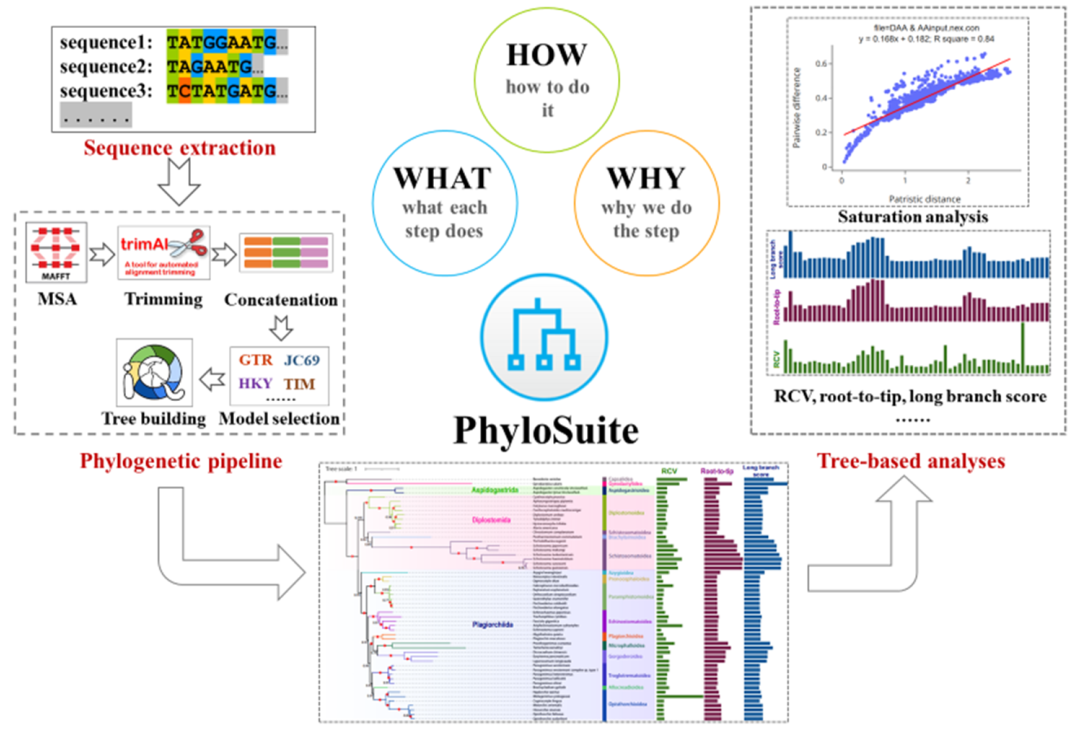
● 本文主要介绍并演示使用 PhyloSuite 进行分子系统发育树重建的所有步骤以及最新开发的11种基于树文件的统计分析功能,包括介绍每一个步骤的背景信息(what)、执行该步骤的原因(why)和具体操作流程(how)。旨在帮助初学者快速入门(多基因联合)系统发育分析,同时提升使用者的分析效率。
● 第一作者:相传钰
● 通讯作者:张东 (dongzhang0725@gmail.com)
● 合作作者:高芳銮、Ivan Jakovlić、雷虹鹏、胡叶、张宏、邹红、王桂堂
● 主要单位:兰州大学生态学院,草种创新与草地农业生态系统全国重点实验室;福建农林大学植物病毒研究所;中国科学院水生生物研究所,农业部淡水养殖病害防治重点实验室,淡水生态与生物技术国家重点实验室
亮 点
● 发布操作更友好、功能更强大的PhyloSuite新版本v1.2.3
● 以“what、why、how”行文结构的PhyloSuite手把手使用教程
● 解决新手入门难、分析效率低等分子系统发育分析常见问题
摘 要
系统发育分析迎来了系统基因组学时代。然而对于系统发育分析的初学者来说,多基因联合系统发育分析门槛较高,且耗时耗力。PhyloSuite是一个整合并流程化了所有多基因联合系统发育分析步骤的可视化分析平台。我们近期发布了PhyloSuite 1.2.3版本,该版本主要对先前版本的bug进行了修复、优化了部分插件(提速)和开发了一些新功能,如去除密码子第三位点建树,以及信噪比计算、饱和度分析等11个基于系统发育树的统计分析。本文主要介绍如何使用PhyloSuite进行分子系统发育树重建并演示上述统计分析新功能,包括介绍每一个步骤的背景信息(what)、执行该步骤的原因(why)和具体操作流程(how)。本文的内容将有助于初学者快速入门(多基因联合)系统发育分析,同时提升使用者的分析效率。
视频解读
Bilibili:https://www.bilibili.com/video/BV1Z24y1V7Uq/
Youtube:https://youtu.be/qtAL8X3314g
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
PhyloSuite分子系统发育树分析教程
https://www.bilibili.com/video/BV1kY411z7Qe/
全文解读
引 言
分子系统发生学旨在利用遗传标记(例如核苷酸和氨基酸序列)重建生物的进化史,被广泛应用于生命科学的各个领域。组学时代的到来推进了分子系统发生学在系统分类、种群遗传、分子定年以及物种适应性进化研究等方面的应用。例如,多基因联合系统发育分析(联合多个基因推导系统发育树)为许多历史遗留的争议问题提供了新的见解,并为我们更好地理解地球上生物的进化历史做出了重要贡献。多基因联合系统发育分析主要包括以下步骤:序列下载及筛选、序列提取、序列比对、序列修剪(可选)、基因串联、最优分区策略和进化模型的选择、系统发育树重建及美化等。除了步骤较多外,每个步骤还有多种软件可以选择。例如,MAFFT、MUSCLE、PRANK等均可用于序列比对,IQ-TREE、RAxML、MrBayes等软件均可用于系统发育树推导。此外,不同软件还往往要求不同的输入和输出文件格式。这无疑加高了初学者的入门门槛,并压缩了科研人员钻研科学问题的时间。
PhyloSuite是一个简单高效的可视化系统发育分析平台,整合并流程化了上述所有分析步骤所需的软件,适用于单基因及多基因联合系统发育分析。PhyloSuite集界面化操作,批量、多核运行,流程化分析等特点于一身(详见引文参考文献[11]),不仅对系统发育分析初学者友好,还可以提高使用者的分析效率。我们近期发布了PhyloSuite新版本(v1.2.3),与1.2.2版本相比主要有以下改进:1. 对一些功能进行了优化,如文件提取、MAFFT密码子比对、序列格式转换、串联和MrBayes等功能提速;2. 对bug进行了修复,如MrBayes中的“内存错误”,运行PhyloSuite时自动更新检查功能报错,在Linux中运行卡死等;3. 开发了一些新功能,如新增11个基于系统发育树的统计分析、新增绘图引擎(适用于RSCU分析、统计分析等功能)、拆分密码子位点建树等(详见https://github.com/dongzhang0725/PhyloSuite/releases/tag/1.2.3)。
基于PhyloSuite新版本,本文将介绍多基因联合系统发育分析的每个步骤的背景信息(what)、执行该步骤的原因(why)以及具体操作方法(how),旨在降低系统发育分析的入门门槛,简化操作流程,并提升使用者的分析效率。
数据集介绍
单基因系统发育分析使用24个纤毛虫(Ciliophora门)的18s基因。多基因系统发育分析使用9个三代虫(Platyhelminthes门,Monogenea纲)的线粒体基因组,包括以下3种数据集:1. 串联所有蛋白质编码基因的核苷酸序列(PCGs)和两个rRNA基因序列(PCGsRNA);2. 串联所有PCGs的密码子第1、2位和两个rRNA基因的序列(PCGs12RNA);3. 串联所有PCGs的氨基酸序列(AA)。
安 装
PhyloSuite的安装教程详见:http://phylosuite.jushengwu.com/dongzhang0725.github.io/installation/或https://dongzhang0725.github.io/installation/。
PhyloSuite界面
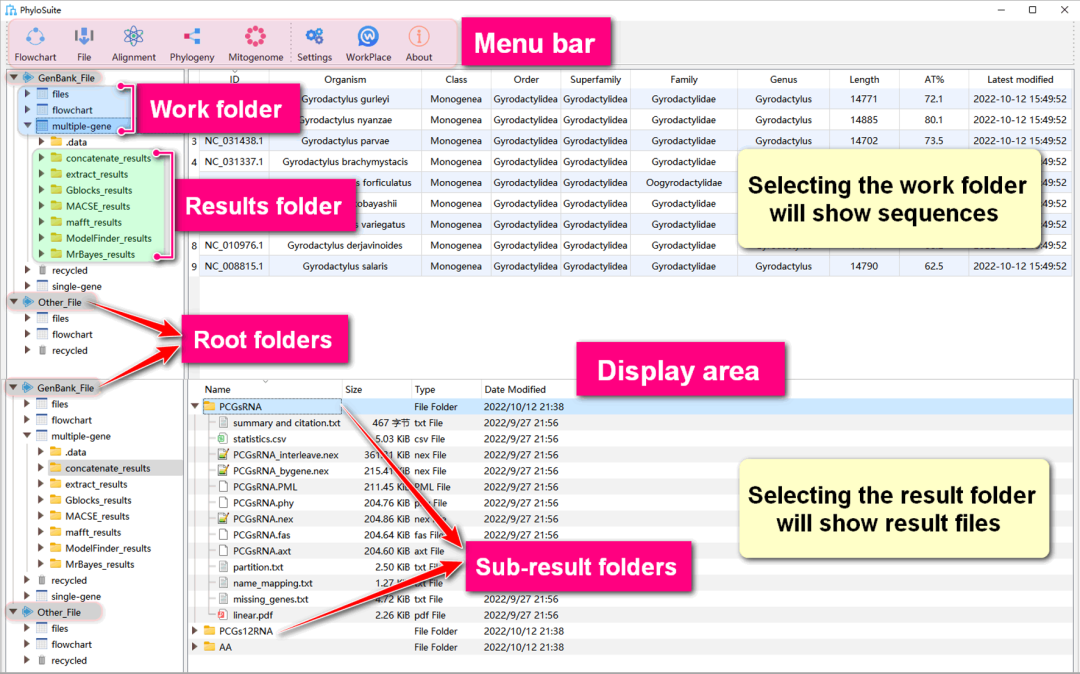
图1. PhyloSuite界面介绍
如图1所示,红色框所示区域为菜单栏,用于选择需要的功能或插件以及设置/更改工作区;灰色框所示区域为根文件夹:“GenBank_File”用于存放GenBank格式的文件和相关输出结果;“Other_File”用于存放其他格式(例如Fasta)的文件和相关输出结果;蓝色框所示区域为工作文件夹;绿色框所示区域为结果文件夹,每个功能都有对应的一级结果文件夹,例如“mafft_results”存放当前工作文件夹下的所有MAFFT分析结果。一级结果文件夹包含多个子文件夹,其名称可以在运行程序之前,点击“Start”按钮右侧的下拉菜单设置,默认名称为当前时间。更多信息详见PhyloSuite手册:http://phylosuite.jushengwu.com/dongzhang0725.github.io/documentation/。
结果
1.多基因联合系统发育
多基因联合系统发育树重建和基于系统发育树的统计分析的教程视频,见http://phylosuite.jushengwu.com/dongzhang0725.github.io/PhyloSuite-demo/videoes/。
1.1 序列下载和准备
线粒体基因组数据通常存储在NCBI的核苷酸数据库(Nucleotide)中,下载步骤如图2所示:
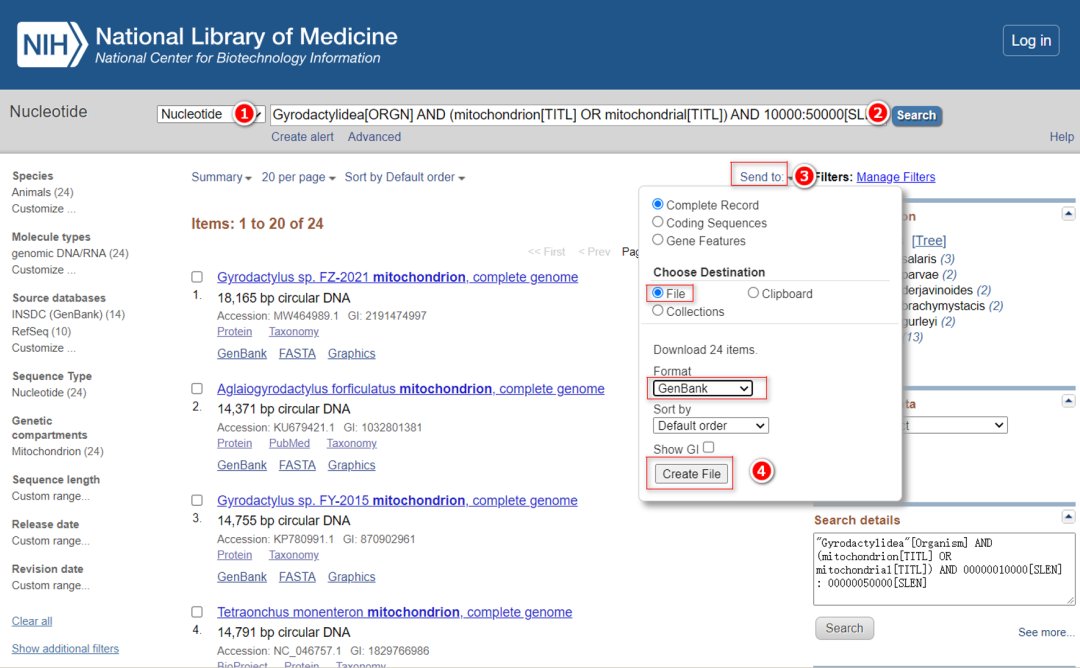
图2. 从NCBI的核苷酸数据库下载线粒体基因组数据
1.1.1. 进入NCBI的官方网站(https://www.ncbi.nlm.nih.gov/),选择“Nucleotide”数据库。注意,节标题最后一级的数字对应图中的红色编号(如节号1.1.1对应图中的红色序号①)。
1.1.2. 在搜索框中输入“Gyrodactylidea[ORGN] AND (mitochondrion[TITL] OR mitochondrial[TITL]) AND 10000:50000[SLEN]”,搜索目标序列。这些关键词可以分为三个部分:第一部分是“Gyrodactylidea[ORGN]”,限制分类名为三代虫目。第二部分是“(mitochondrion[TITL] OR mitochondrial[TITL])”,限制序列类型为线粒体基因组。最后一部分(10000:50000[SLEN])将序列长度范围限制为10,000-50,000个碱基。这些参数均可根据需要自行修改。
1.1.3. 选择“send to”保存所有序列。
1.1.4. 依次选择“Complete Record” “File” “GenBank” “Create File”将序列保存为GenBank格式文件。
1.2 将序列导入PhyloSuite
导入序列之前,在PhyloSuite中创建一个新的工作文件夹,用于存储数据和结果。
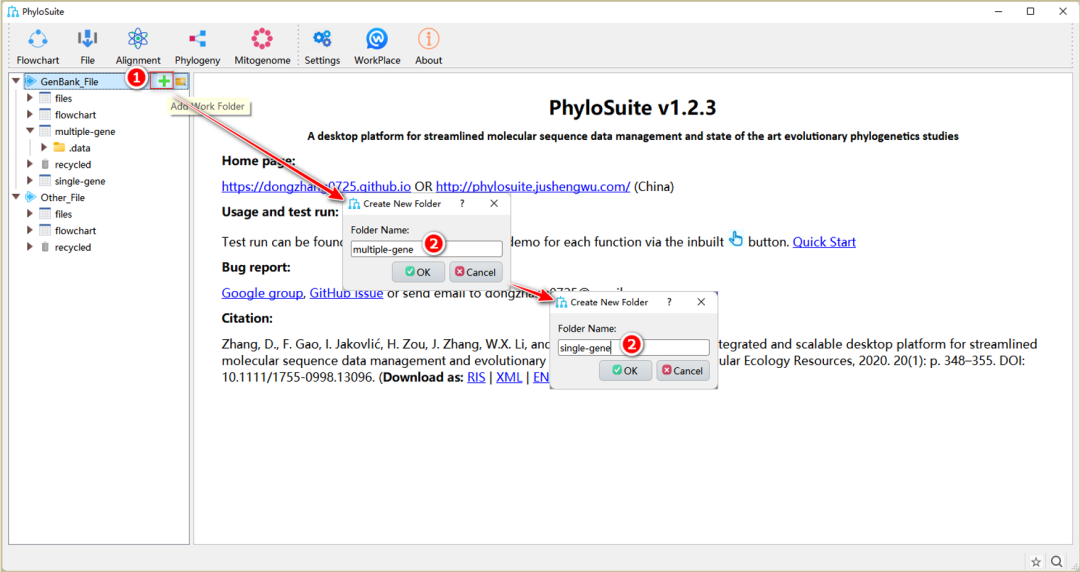
图3. 在PhyloSuite中创建新的工作文件夹
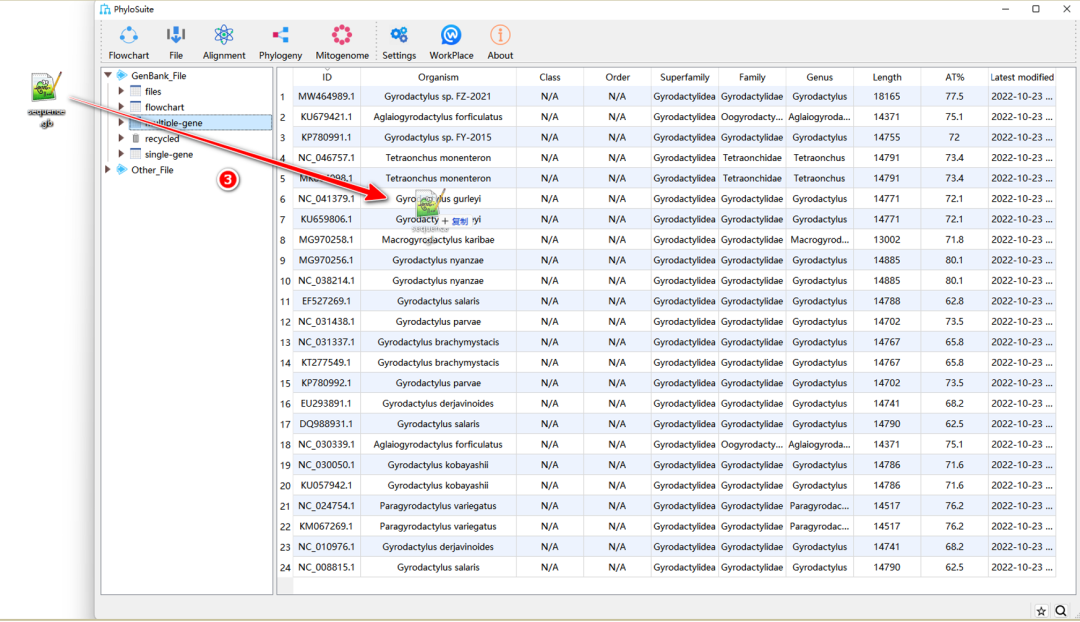
图4. 将基因序列导入PhyloSuite工作文件夹
1.2.1. 将鼠标悬停于GenBank_File根文件夹,然后点击右侧的“+”按钮,创建一个新文件夹并命名(图3)。注:“GenBank_File”根文件夹下的任一文件夹都可作为工作文件夹(例如“files”文件夹)。
1.2.2. 多基因系统发育分析的工作文件夹命名为“multiple-gene”(图3)。
1.2.3. 将步骤1.1.4中下载的序列文件(*.gb)拖到PhyloSuite界面区域(图4所示的区域),导入序列。
1.3. 删除冗余序列
如果线粒体基因组已经通过了RefSeq数据库筛选,通常会有两个登录号,因此为避免序列重复,需要在开始下游分析之前过滤冗余序列。操作如图5所示:
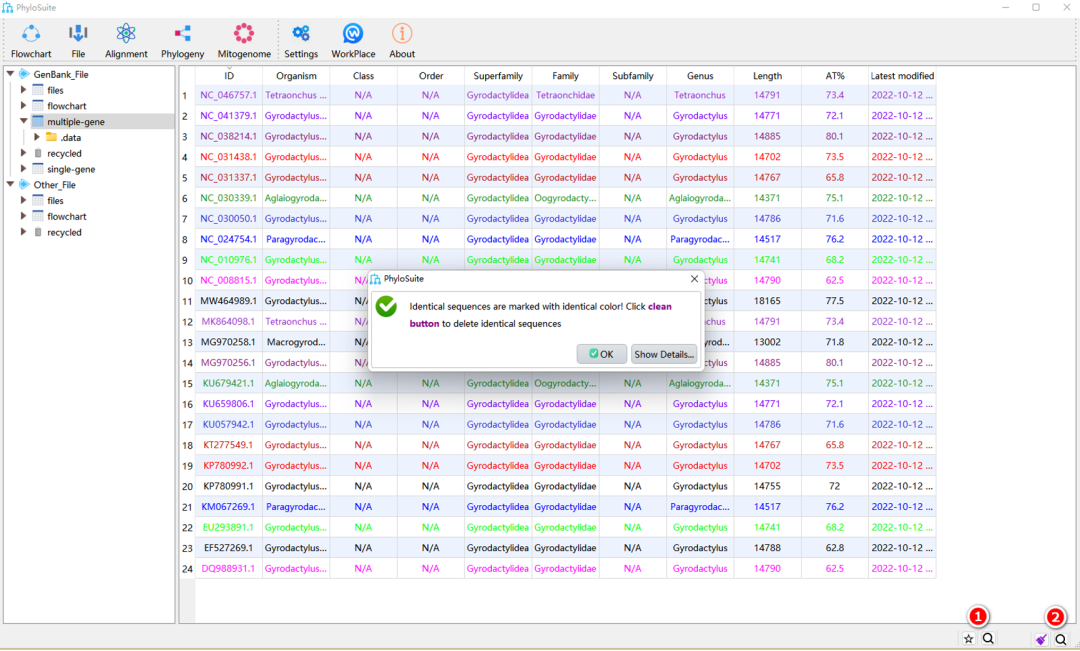
图5. 过滤冗余序列
1.3.1. 成功导入序列后,单击界面右下角的星号按钮。将出现一个如图5所示的消息框,提示相同的序列已用相同的颜色标记。
1.3.2. 五角星变成了扫帚图标。单击后冗余序列将被清除,登录号以NC开头的序列将被优先保留。此外,还可以根据需要手动删除序列(例如物种名重复、序列错误、没有注释信息的序列等):选择要删除的序列,单击右键后选择“delete”。最后注意确认序列中是否保留了外群和自己的序列(常见错误)。
序列导入后,其分类信息可能缺少或错误,PhyloSuite可以从NCBI数据库或WORMS数据库中获取最新的分类信息,操作如下:全选序列后单击右键,选择“Get taxonomy (NCBI, fast)”或“Get taxonomy (WoRMS, slow)”从NCBI或WORMS数据库中获取分类信息(图6)。此外,还可以双击表格单元格手动编辑分类信息。
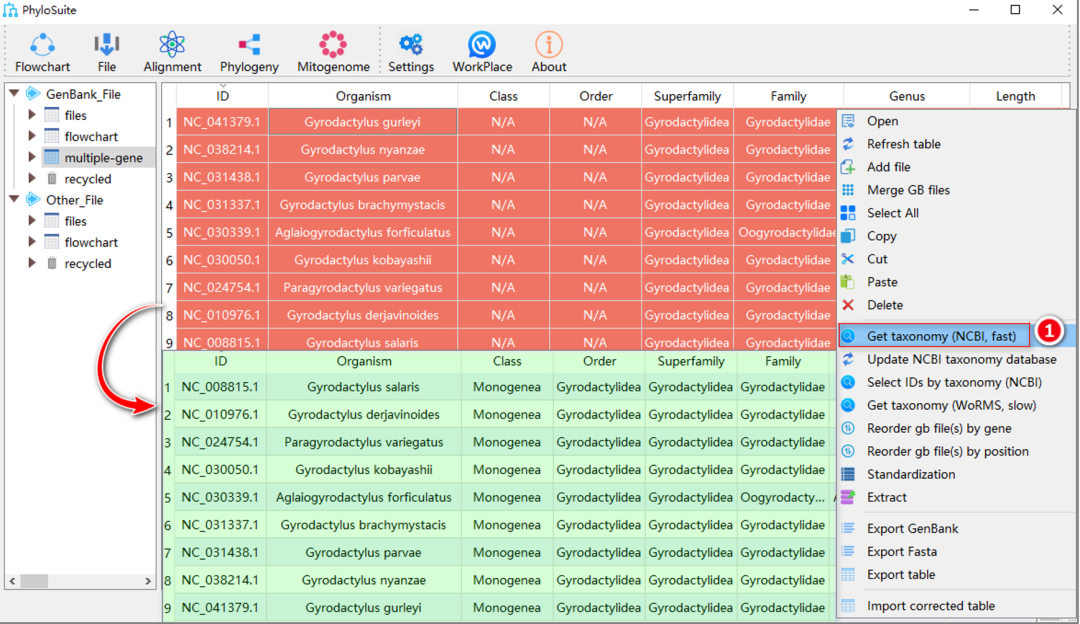
图6. 获取分类信息
1.4. 序列提取
标准的动物线粒体基因组通常包含12或13个PCG,22个tRNA和2个rRNA基因。PhyloSuite可以将它们提取出来用于下游分析,该功能也适用于其他类型的分子数据,如单基因序列、叶绿体基因组、质粒基因组、细菌基因组和病毒基因组等。提取步骤如图7所示。
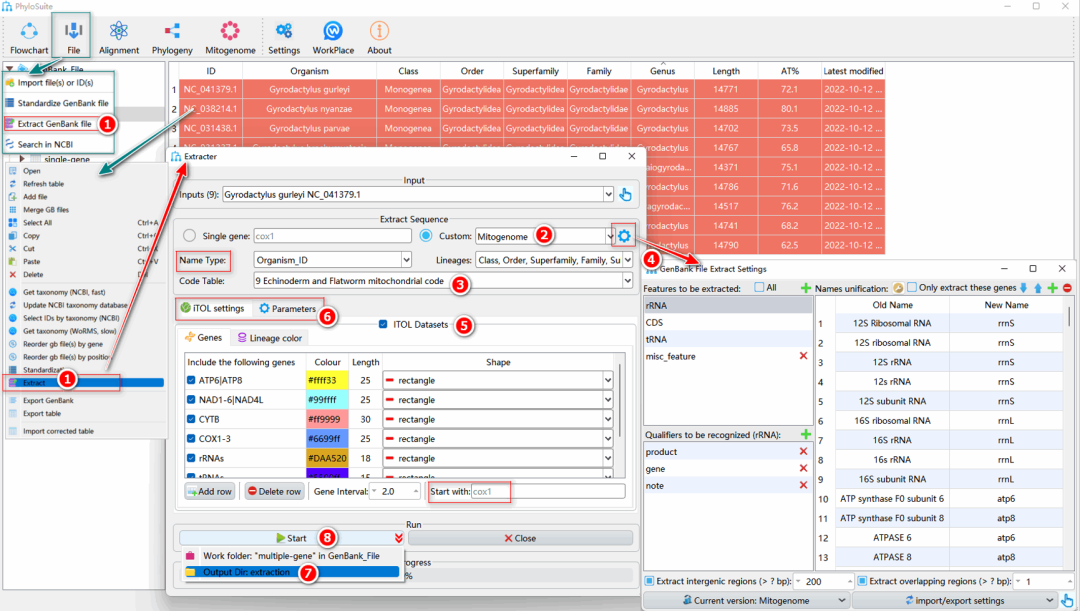
图7. 从线粒体基因组中提取基因序列
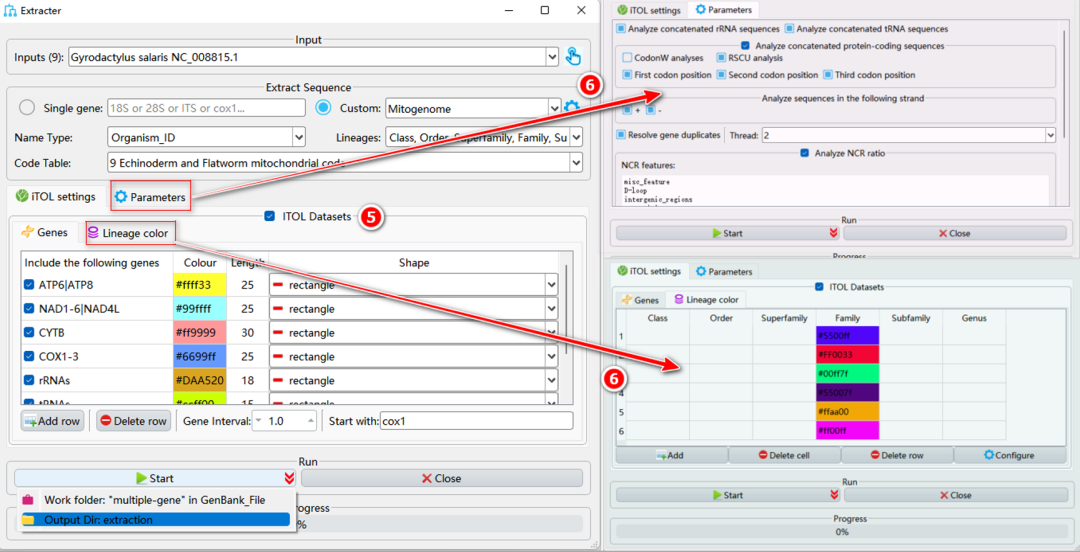
图8. 提取步骤的参数设置
1.4.1. 按Ctrl+A全选序列,单击右键后选择“Extract”(或点击“File”–“Extract GenBank file”),弹出“Extracter”窗口。
1.4.2. 从“Custom”下拉菜单中选择与数据匹配的提取模式。此处选择“Mitogenome”。
1.4.3. 密码表:在“Code Table”的下拉菜单中选择与数据匹配的密码表,以正确识别终止密码子和正确翻译基因序列。Gyrodactylidea的线粒体基因组使用第9套密码表(棘皮动物和扁形动物门线粒体密码表)。
1.4.4. 统一基因别名。提取功能主要通过基因名称来识别同源基因,但同一基因通常在不同数据集中以不同的名称注释:例如,COX1、COI和COXI都是同一种线粒体基因的常用名称,因此提取前需先设置将基因名称统一。单击“Extracter”窗口中“Custom”选项右侧的齿轮形按钮,弹出设置窗口。在提取过程中,“Old Name”列中的名称都将被替换为“New Name”列中相应的名称。PhyloSuite内置了一些常见的基因别名,如需添加新的别名,单击上方的“+”按钮添加新行,或点击“Import”按钮导入别名设置表(见下文)。如数据集包含许多默认设置中未包含的基因名称,推荐先使用“general”模式提取数据,然后打开“StatFiles”文件夹并找到“name_for_unification.tsv”文件。在此文件中设置好“New Name”列中所有基因别名后,单击“Import”按钮将其导入到设置窗口。关于自定义提取的详细教程,见http://phylosuite.jushengwu.com/dongzhang0725.github.io/PhyloSuite-demo/customize_extraction/。
有关“Extract Sequence”参数框内的其他设置:
a. Name Type:自定义序列名称。
b. Lineages:下拉菜单选择在结果中想要显示的分类信息(用于统计表,iTOL文件等)。上述所有步骤的设置见图7。
1.4.5. 用于iTOL系统发育树美化和其他选项的参数设置如图8所示。
a. Genes标签页:设置希望在iTOL中可视化的基因的颜色、长度和形状。通过“Start with”输入框设置基因顺序图的起始基因。
b. Lineage color标签页:双击单元格以设置分类名称的颜色。如果设置的颜色数量少于分类名称的数量,剩余的分类名将被随机分配颜色。例如,共10个分类名,只设置了5种颜色,其余5个分类名将随机分配颜色。
1.4.6. Parameters标签页:选择需要进行的分析。
1.4.7. 单击“Start”按钮右侧的箭头后,单击“Output Dir”以设置输出文件夹的名称(这里命名为“extraction”,后续分析都会以同样的方式设置输出文件夹名称,并不再赘述)。
1.4.8. 最后,单击“Start”按钮开始提取基因序列。
1.5. 多重序列比对
什么是多重序列比对?
多重序列比对(multiple sequence alignment,MSA)是通过推导序列之间匹配、替换和插入(或缺失)碱基的位置,从而鉴定序列之间的同源区域的过程。此过程通常会通过在序列中填充空缺(gaps,用“-”表示),使所有序列长度相等(图9)。
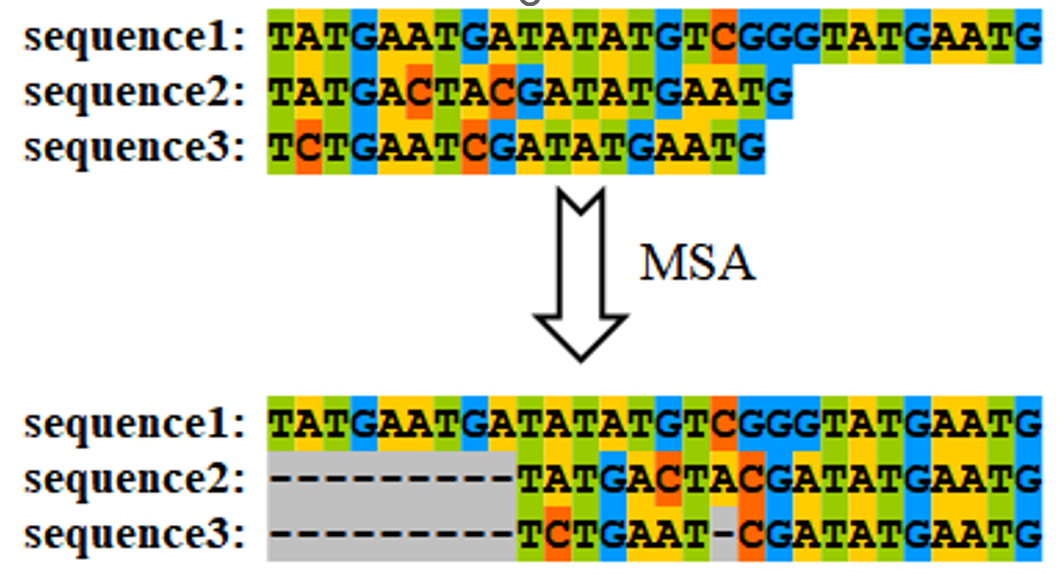
图9. 多重序列比对图示,空缺(gaps)用“-”表示
为什么要进行MSA?
序列的同源关系是系统发育分析的基础,因此MSA是关键的步骤。错误的MSA将对下游分析(例如系统发育,同源建模,数据库搜索,模体(motif)搜寻,基因组注释等)的准确性产生负面影响。
为什么用MAFFT进行MSA?
MSA分析主要考虑速度和准确性两方面因素。在Clustalw,Clustal Omega,DIAIGN-TX,MAFFT,MUSCLE,Poa,Probalign,Probcons和T-coffee等常见的MSA程序中,MAFFT在准确性和速度方面的综合表现优于其它软件,尤其是处理大数据集。MAFFT的simplified scoring system算法不仅可减少CPU耗时,还可提高基因序列的比对精准度,尤其是对于具有大量插入、缺失,且相似度较低的序列。
如何在PhyloSuite中使用MAFFT?
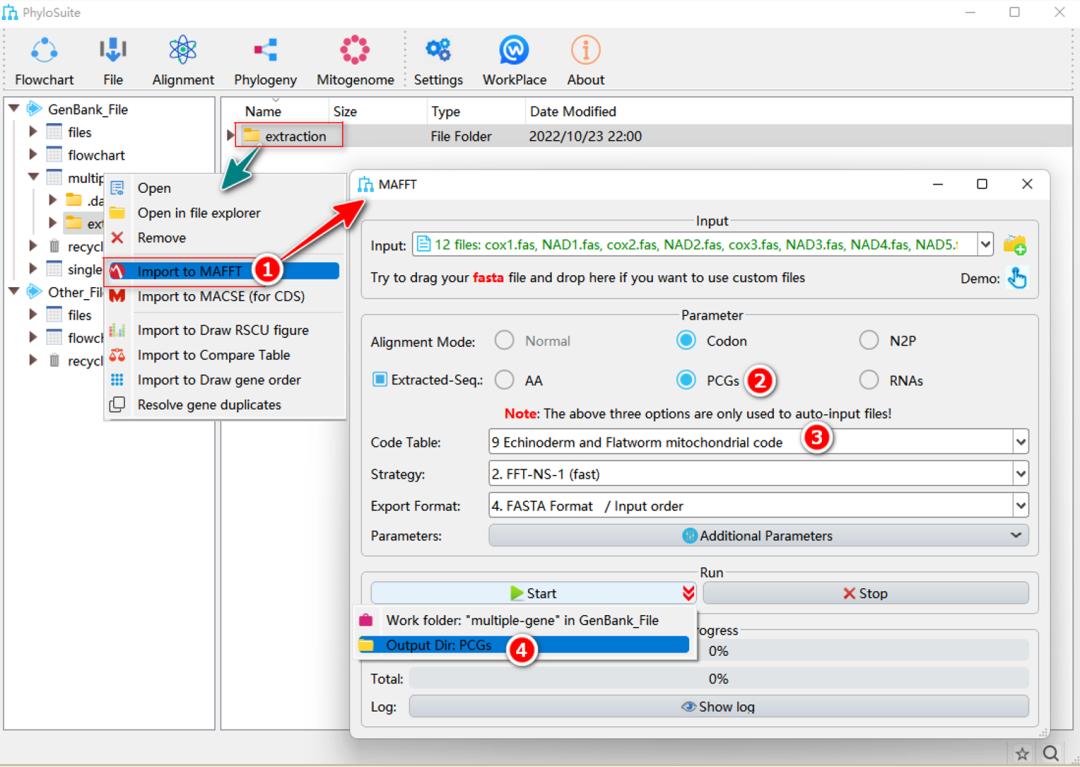
图10. 使用MAFFT进行多重序列比对
1.5.1. 右键单击“Extraction”的结果文件夹,然后选择“Import to MAFFT”。
1.5.2. 提取结果导入MAFFT时,将自动勾选“Extracted-Seq”复选框,共有三种模式(AA、PCGs和RNA)可以选择:选择“PCGs”将自动导入提取的PCGs核苷酸序列(同时勾选“Codon”比对模式);选择“AA”将自动导入提取的PCGs氨基酸序列(同时勾选“Normal”模式);选择“RNA”将自动导入所有tRNA和rRNA基因的序列(同时勾选“Normal”模式)。
注意,仅导入PhyloSuite的提取结果到MAFFT时,以上三种模式才会启用。“Codon”比对模式属于PhyloSuite为MAFFT新增的功能,其原理为:先将核苷酸序列翻译为氨基酸序列,MAFFT比对后再回译为相应的三联体密码子核苷酸序列。
1.5.3. 选择Gyrodactylidea对应的第9套密码表(该选项仅适用于“PCGs”)。
1.5.4. 设置结果文件夹名称(这里不同数据的结果文件夹分别命名为“PCGs”,“AA”和“RNAs”),然后单击“Start”按钮开始多重序列比对(图10)。
如何在PhyloSuite中比对外源序列(即不在PhyloSuite工作区的文件)?
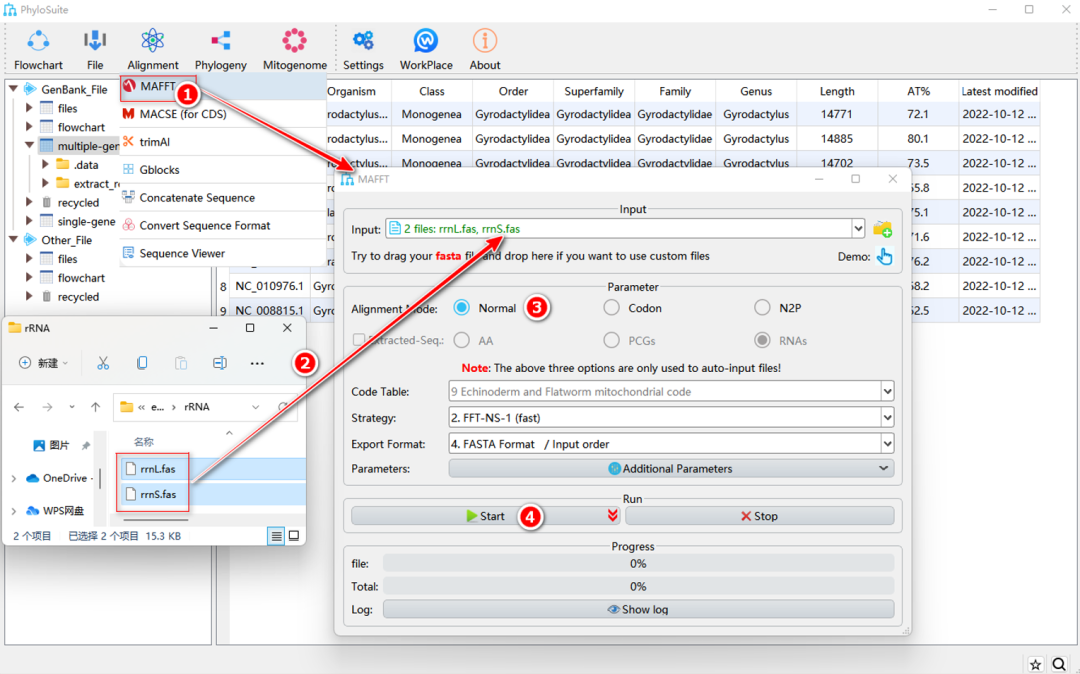
图11. 在MAFFT中比对外源序列
将“Fasta”格式的外源基因序列文件拖到“Input”输入框中,在“Alignment Mode”中选择适用于当前数据的选项。注意,此时“Extracted-Seq”中的选项将被禁用。其他步骤与1.5.2-1.5.4中相同(图11)。
MAFFT的其他参数可以根据需要设置,详见官方文档https://mafft.cbrc.jp/alignment/software/manual/manual.html。
1.6. 使用MACSE优化PCGs比对(可选)
为什么使用MACSE优化PCGs比对?
PhyloSuite中,MAFFT的“Codon”比对模式与MEGA和TranslatorX的原理类似,未考虑PCGs的密码子结构,当提早产生终止密码或存在移码突变时会产生比对错误。与MAFFT等不同,MACSE使用经典的“Needleman-Wunsch”算法,能正确识别假基因化事件,并保持祖先密码子结构。由于直接使用MACSE比对PCGs序列的速度过慢(尤其是大数据),因此本教程将演示先使用MAFFT进行PCGs序列比对,然后使用MACSE进行优化。操作步骤如图12所示。
如何在PhyloSuite中使用MACSE?
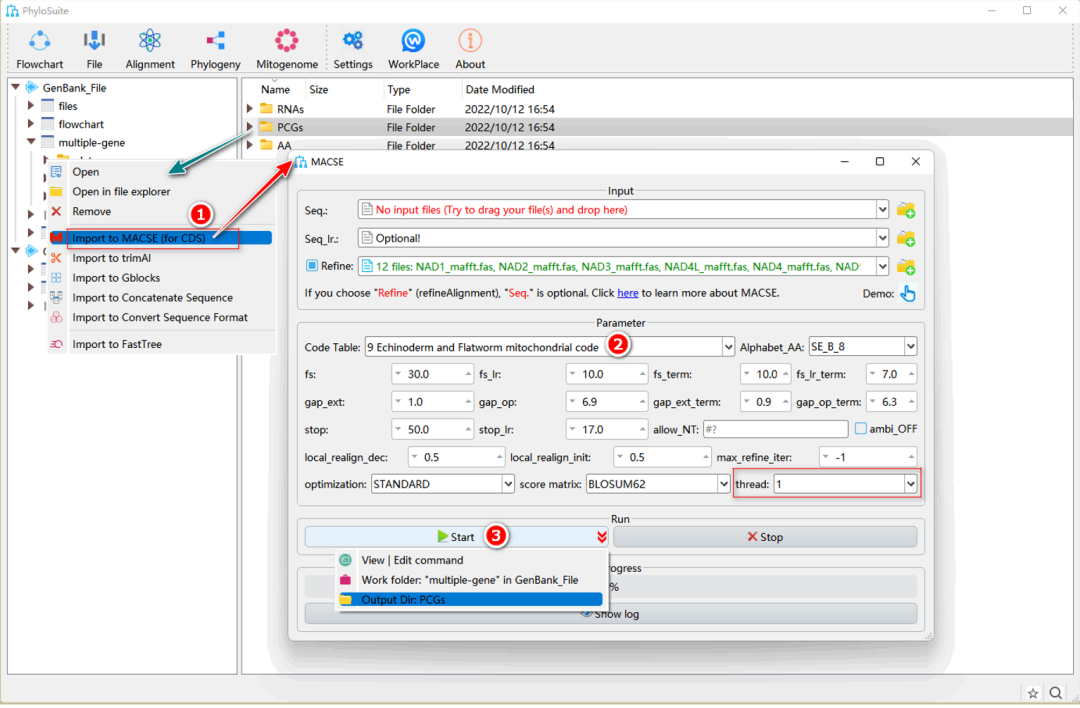
图12. 使用MACSE优化蛋白编码基因序列比对结果
1.6.1. 右键单击“PCGs”的“MAFFT”结果文件夹,然后选择“Import to MACSE (for CDS)”。此时“Refine”框自动选中,MAFFT比对结果被自动导入。
1.6.2. 选择Gyrodactylidea对应的第9套密码表。
1.6.3. 设置结果文件夹名称(在这里命名为“PCGs”),然后单击“Start”按钮开始运行。
注意,MACSE的结果文件中,检测到的移码突变将会用“!”或“*”符号标记。由于这些符号可能会影响下游分析,因此除了MACSE的原始输出文件(文件名中带有“_nt”和“_aa”字样)之外,PhyloSuite还会生成文件名中带有“removed_chars”字样的文件,主要将上述特殊字符替换为“?”,从而方便下游分析。
如何使用外源序列进行MACSE分析?
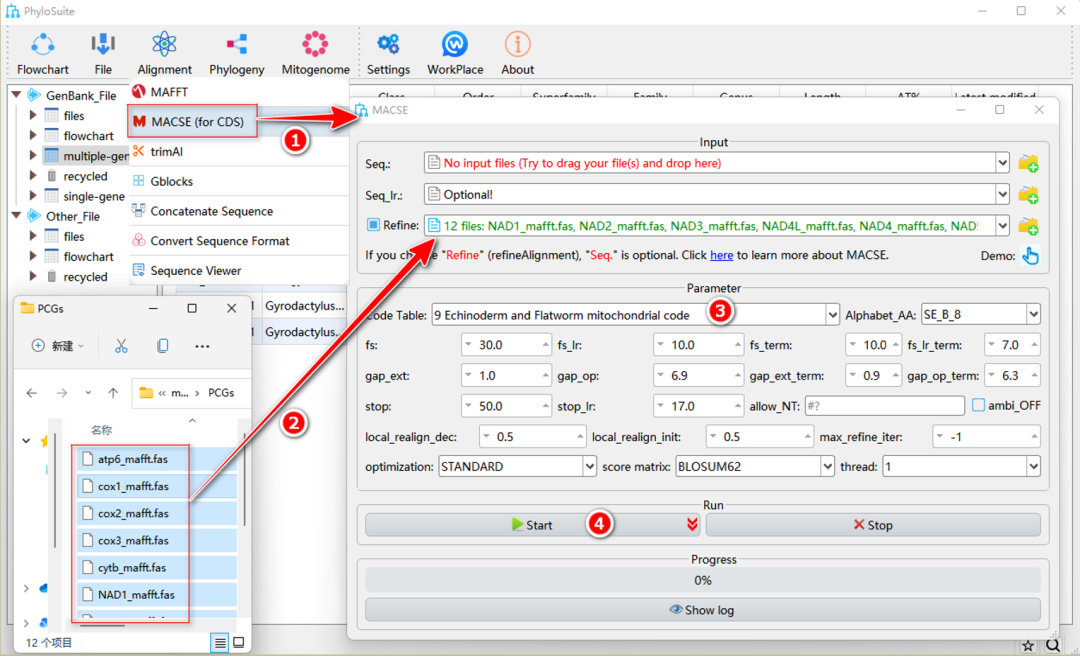
图13. 使用外源基因序列在MACSE中比对
选择准备好的“Fasta”格式的PCGs基因序列文件,将它们拖入“Seq”输入框(适用于未比对的序列)或“Refine”输入框(适用于已比对的序列)(图13)。其他步骤与1.6.2-1.6.3中相同。
注意,MACSE只能用于蛋白编码基因核苷酸序列的比对。MACSE的其他参数可自行设置,详见https://bioweb.supagro.inra.fr/macse/index.php?menu=intro。
1.7. 序列修剪(可选)
什么是序列修剪?
序列修剪是指去除低质量比对位点,包括MSA过程中错误推导的同源区域或位点,以及存在大量插入或缺失的区域(图14)。
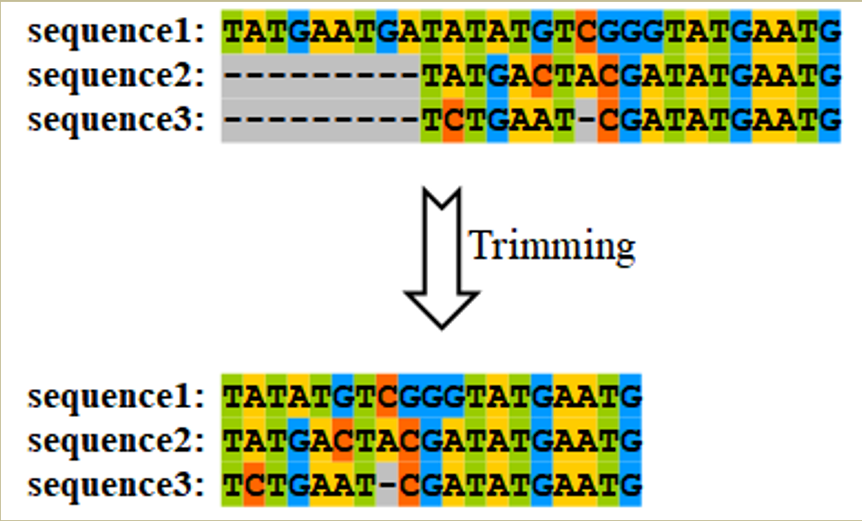
图14. 序列修剪的图示
为什么修剪序列?
序列比对的准确性对下游系统发育分析至关重要,但目前常用的算法均无法将基因序列完美比对。修剪序列可以去除存在比对错误,或是多重替换的位点,以达到去除系统发育噪声和保留系统发育信号的目的,进而提高下游分析的准确性和速度。另外,一些进化模型会将空缺(gaps)视为第五种碱基。
为什么使用Gblocks进行PCGs序列修剪?
HmmCleaner(仅在PhyloSuite的Linux版本中可用,适用于AA序列)使用的segment-filtering法在进化事件推导方面优于Gblocks和trimAl使用的block-filtering法。而对于使用block-filtering法的两个软件而言,数据量较大时trimAl优于Gblocks。PCGs序列需要使用Gblocks,因为它的“Codon”模式可以以三联体密码子形式进行序列修剪;RNAs序列建议使用trimAl,AA序列建议使用trimAl或HmmCleaner。
如何在PhyloSuite中进行序列修剪?
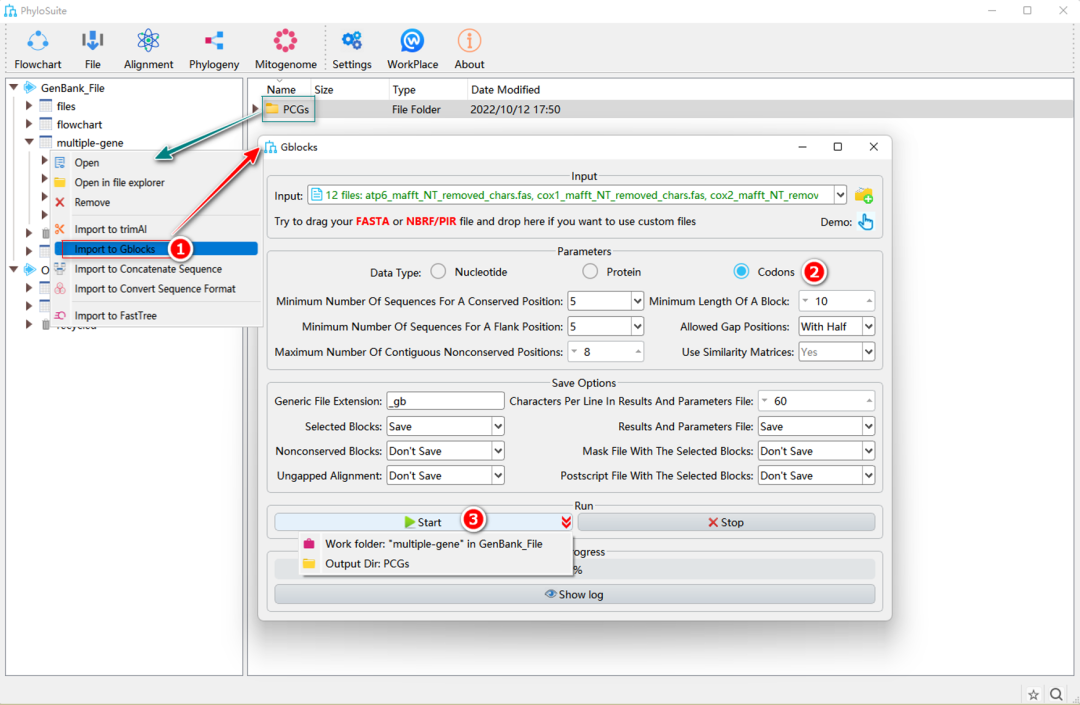
图15. 使用Gblocks修剪PCGs序列
1.7.1.1. 右键单击MACSE(PCGs)或MAFFT(RNAs和AA)的结果文件夹,然后选择“Import to Gblocks”。
1.7.1.2. 选择数据集对应的数据类型,PCGs选择“Codons”,RNAs选择“Nucleotide”,AA选择“Protein”。
1.7.1.3. 设置结果文件夹名称(此处命名为“PCGs”),然后单击“Start”按钮开始运行(图15)。
如何修剪RNA序列?
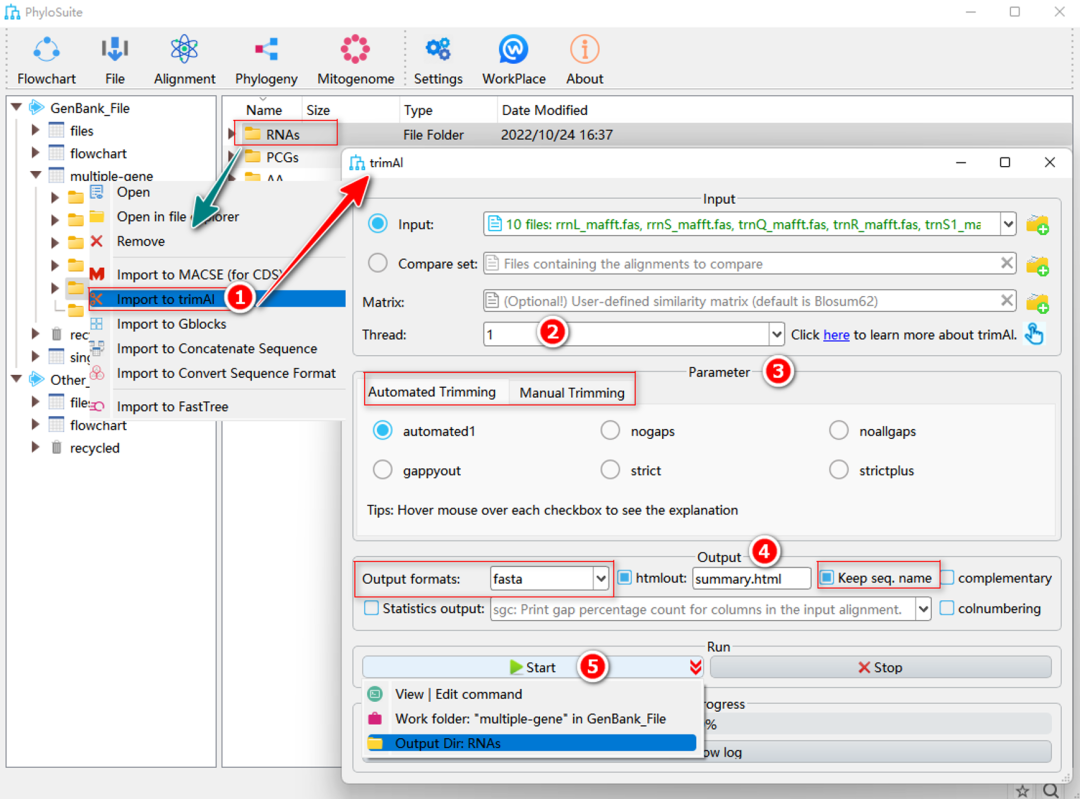
图16. 使用trimAl修剪RNA序列
1.7.2.1. 右键单击RNA基因的MAFFT结果文件夹,然后选择“Import to trimAl”。
1.7.2.2. “Threads”参数可用于并行修剪多个文件。
1.7.2.3. 选择合适的修剪参数。
a. Manual Trimming:自定义修剪参数,例如gaps阈值。
b. Automated Trimming:根据user-defined(“nogaps”或“noallgaps”)或MSA features-based(“gappyout”,“strict”和“strictplus”等方法)设置的gaps阈值进行修剪。“automated1”是一种启发式方法,可自动选择最优方法进行修剪。trimAl的其他参数设置详见http://trimAl.cgenomics.org/_media/manual.b.pdf.
1.7.2.4. 在“Output”中设置参数。
a. “Output formats”:设置输出文件格式。
b. “Keep seq. name”:确保序列名称不被精简。
1.7.2.5. 设置结果文件夹名称(在这里命名为“RNAs”),然后单击“Start”按钮开始修剪(图16)。
如何修剪外源序列?
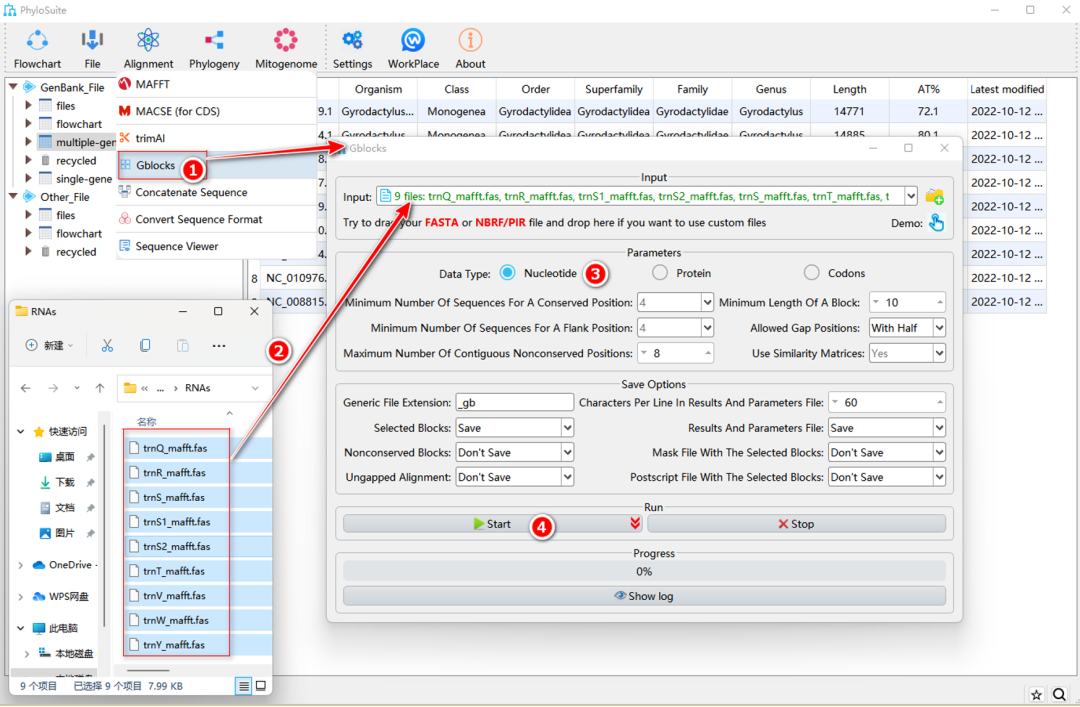
图17. 使用Gblocks修剪外源序列
将准备好的“Fasta”格式基因序列文件拖到Gblocks的“Input”输入框中,然后选择相应的“Data Type”(图17)。其余步骤与1.7.1.2-1.7.1.3中相同。
注意,并非所有基因序列都需要修剪,因为修剪存在“得”与“失”的权衡问题,目前修剪MSA的程序可能并不能完美修剪“有问题”的位点,甚至有时会导致一些有用的信息位点丢失。Gblocks其他参数设置详见http://molevol.cmima.csic.es/castresana/Gblocks/Gblocks_documentation.html。
1.8. 序列串联
什么是序列串联?
序列串联是指将比对完成的多个单基因序列首尾相接连成“超矩阵”。串联时主要通过序列的名称来识别,如三个基因文件中名为“Species1”的序列将会被串连在一起(图18)。缺失基因的位点用“?”表示。
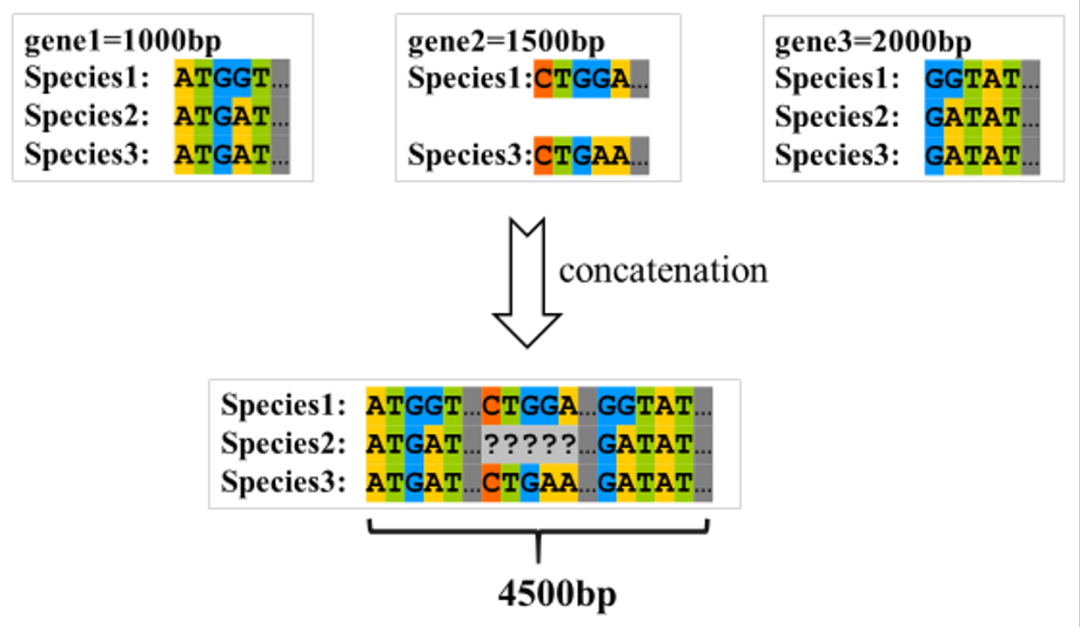
图18. 序列串联的示意图
为什么串联序列?
多基因串联后的序列通常比单基因携带更多系统发育信息,更容易生成拓扑结构单一且支持率较高的系统发育树。
如何在PhyloSuite中串联序列?
PCGsRNA数据集生成过程如图19所示:
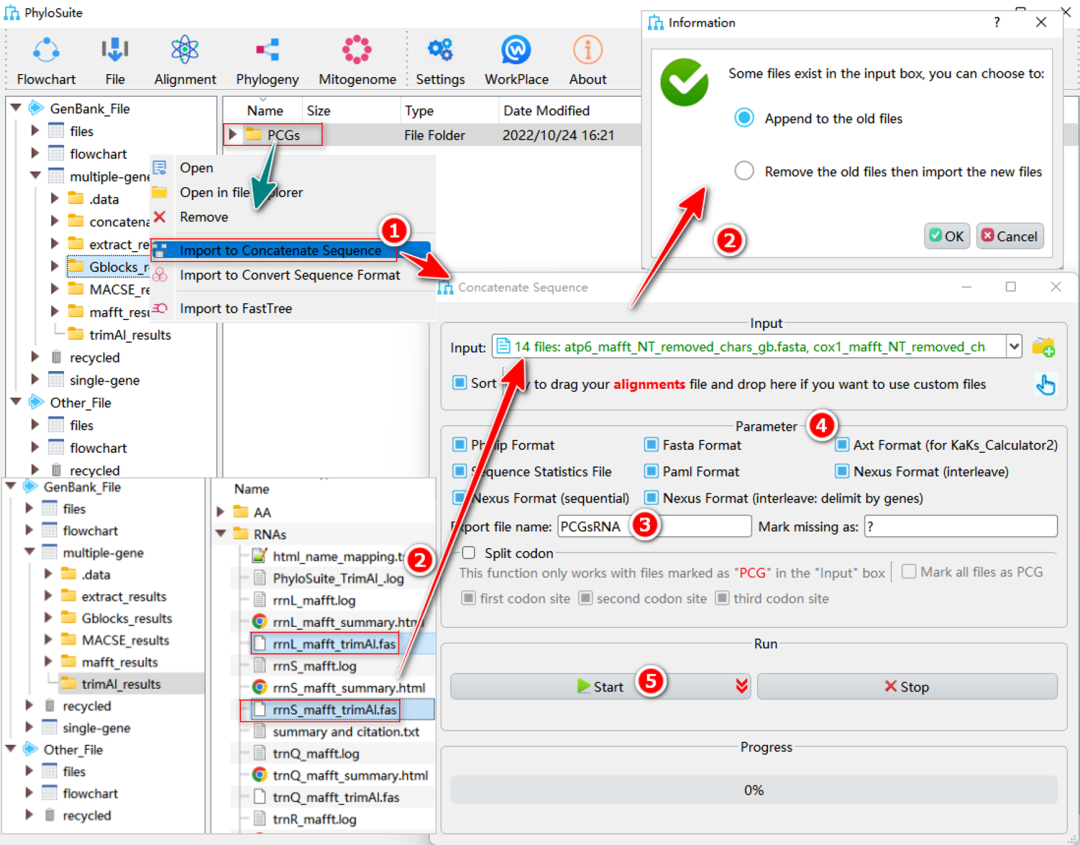
图19. PCGsRNA数据集的多序列串联
1.8.1. 右键单击PCGs的“Gblocks”结果文件夹,然后选择“Import to Concatenate Sequences”,修剪完成的PCGs将自动导入到“Input”输入框中。
1.8.2. 打开RNA基因的“trimAl”结果文件夹,选择修剪完成的序列文件(名称中带有 “*_trimAl.fas”)拖到“Concatenation”界面的“Input”输入框中。选择“Append to old files”选项将RNA文件追加到已导入的PCGs文件中。
1.8.3. 在“Export file name”中设置输出文件的名称。
1.8.4. 在“Parameter”中选择文件的输出格式。
1.8.5. 设置结果文件夹名称(在这里命名为“PCGsRNA”),然后单击“Start”按钮开始串联序列。
如何串联生成PCGs12RNA数据集(去除第三位密码子位点)?
在远缘物种的数据集中,PCGs的第3位密码子由于进化速度较其它位点快,通常会导致序列替换饱和,进而阻碍正确的系统发育关系推导。因此对于此类数据集,研究者们通常会选择将PCGs数据集的第3位密码子去掉,以得到更加合理的系统发育结果。具体操作如图20所示。
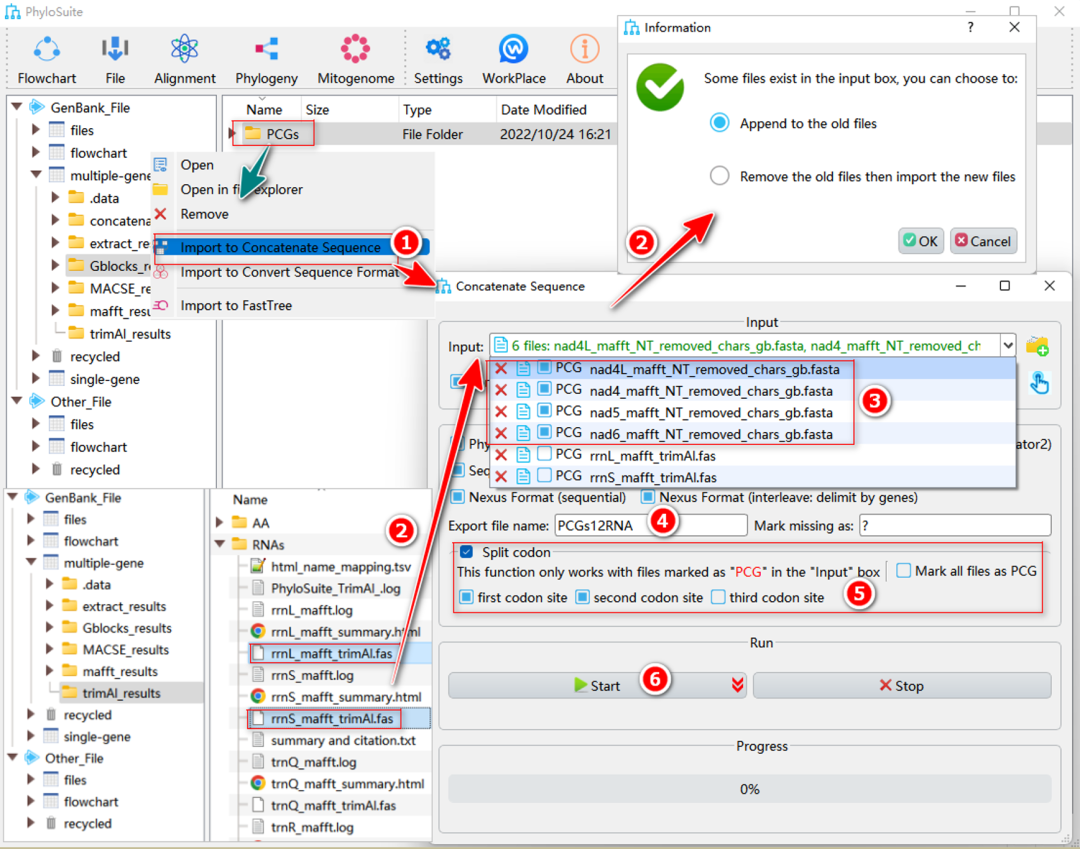
图20. 串联生成PCGs12RNA数据集
序列导入步骤与1.8.1和1.8.2节中所述相同。导入后,单击“Input”输入框弹出文件列表,将列表中蛋白编码基因文件对应的“PCG”选框勾选。如果PCGs文件较多,可以使用“Mark all files as PCG”选项将所有输入文件先勾选,再将非PCGs文件取消勾选(此处为了方便演示,只串联了六个文件)。然后,选中“Split codon”选框,同时勾选“first codon site”和“second codon site”选项(“third codon site”保持非勾选状态)。其他操作与1.8.2-1.8.5的步骤类似(输出文件和文件夹均命名为“PCGs12RNA”)。
如何串联生成AA数据集?
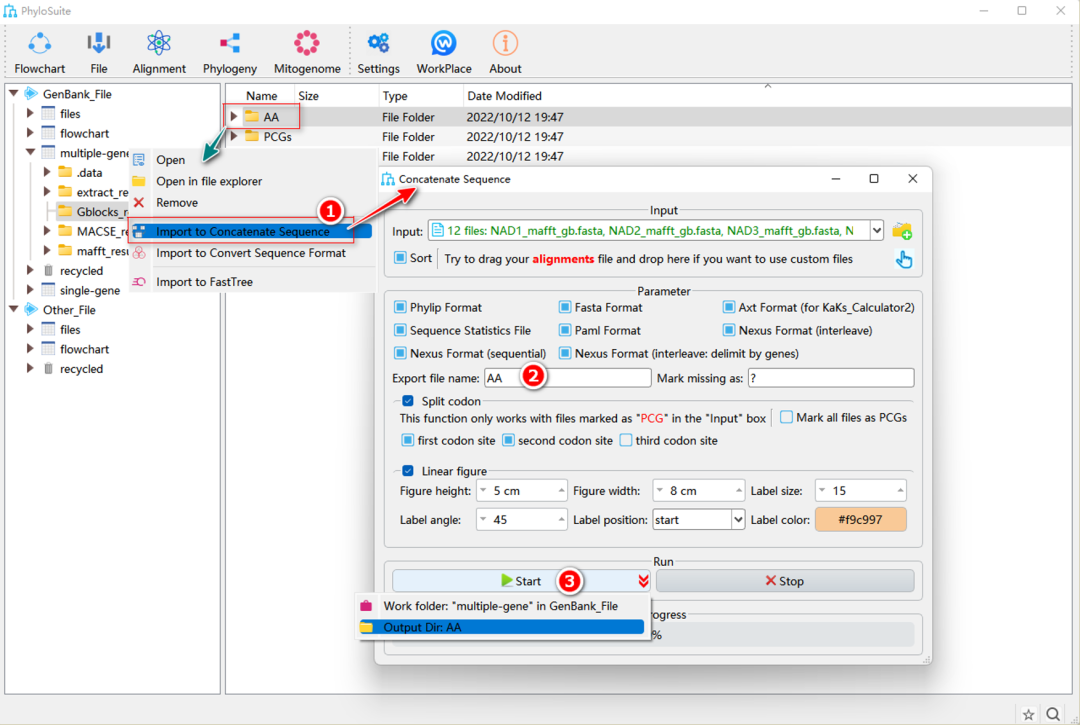
图21. 串联生成AA数据集
右键单击AA的“Gblocks”或“trimAl”结果文件夹,选择“Import to Concatenate Sequences”(图21)。其他步骤与1.8.3-1.8.5节相同。
如何串联外源比对序列?
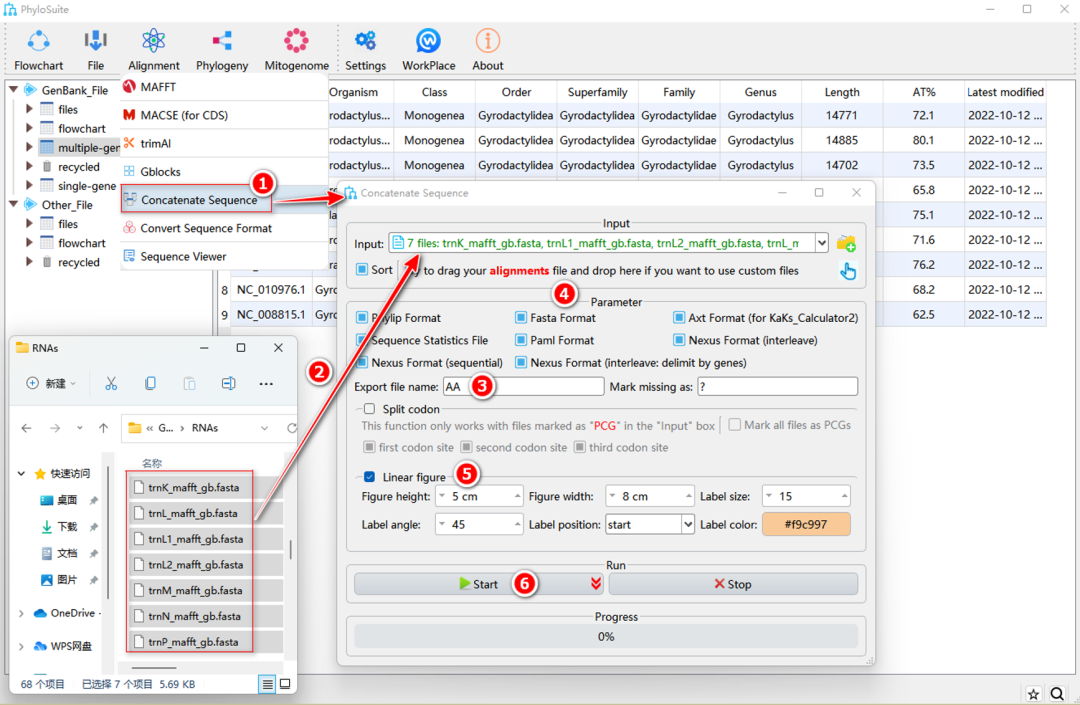
图22. 外源比对序列的串联
选择准备好的序列文件(“Fasta”,“Phylip”或“Nexus”格式),拖到“Input”输入框中(图22)。后续步骤与1.8.3-1.8.5相同。
1.9. 最优分区策略和模型选择
若不分区直接选择最优进化模型,见附件中的第5.4节“最优进化模型选择”,然后直接跳转到本文的第1.10节。
什么是分区?
分区(partitioning)是指将序列中不同基因或位点划分为不同亚集(subset),然后分别估计它们的进化模型。例如,我们可以将PCGsRNA数据集中的12个PCGs和2个rRNA基因视为单独的分区(14个预分区),然后将具有相似进化过程的预分区组合为亚集(即确定最优分区策略),并为每个亚集选择最优进化模型。注意,每个蛋白编码基因还可以根据密码子位点进一步拆分,因此12个PCGs还可拆分为36个预分区(见下方操作步骤)。
为什么要进行分区?
不同基因通常执行不同的生物学功能,因此可能受到不同的选择压力,进而导致进化速率也不同。因此,如果多基因串联的数据集都使用同一个模型(即假设所有基因或位点都具有相同的进化过程)可能会引起系统发育关系推导错误。选择最优分区策略(将具有相似进化过程的基因或位点组合为一个亚集),并分别为每个亚集选择最优进化模型,可以有效解决以上问题(详见引文参考文献[36, 38])。
如何在PhyloSuite中选择最优分区策略和进化模型?
使用ModelFinder为PCGsRNA数据集选择最优分区策略和进化模型的操作步骤如图23所示。
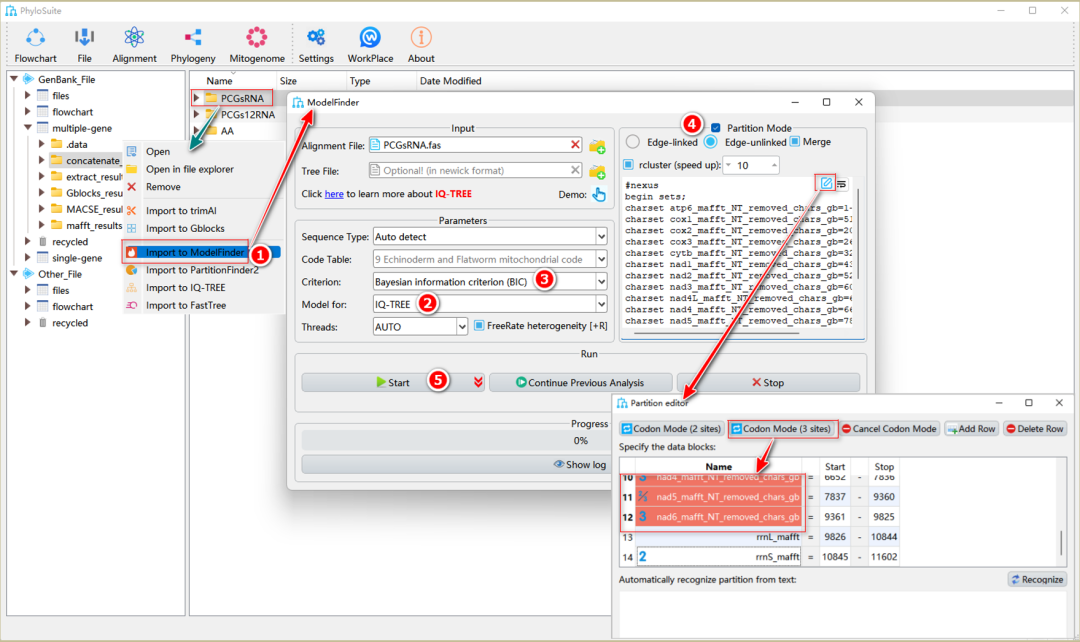
图23. 使用ModelFinder进行分区分析
1.9.1.1. 选择PCGsRNA的“Concatenation”结果文件夹,单击右键并选择“Import to ModelFinder”。修剪完成的序列和分区详细信息将被自动导入“Input”和“Partition Mode”输入框中。
1.9.1.2. “Model for”:选择下游建树软件。
1.9.1.3. “Criterion”:选择用于判断模型适合度的标准。默认为BIC,值越小代表模型越适合。
1.9.1.4. “Partition Mode”:
a. Edge-linked/unlinked:“Edge-linked”允许每个分区有各自的进化速率,但是枝长相同。当需要评估数据的heterotachy的时候,参数丰富但速度较慢的“Edge-unlinked”选项更适合,它允许各分区拥有各自的枝长。注意,使用MrBayes建树时,“Edge-unlinked”参数会导致每个分区生成一棵独立的系统发育树。
b. Merge:合并具有相似进化过程的分区,从而避免过度参数化并增强模型拟合度。
c. rcluster:指定使用relaxed clustering算法评估分区方案的比例,以减少计算负担、加快分析速度。例如,10代表只评估前10%的分区方案。
d. Partition data block edit:单击分区编辑框右上角的铅笔形按钮以编辑分区。步骤如下:选择所有PCGs分区,然后单击顶部的“Codon Mode (3 sites)”按钮,每个蛋白编码基因的分区将会被拆分为3个密码子分区,其中图标1、2和3分别代表相应蛋白编码基因的第1、2和3密码子位点的分区。注意非PCGs数据不能用“Codon Mode”功能。
1.9.1.5. 设置结果文件夹名称(这里命名为“PCGsRNA”),然后单击“Start”按钮开始运行。
提示:在结果文件夹中,最优模型分区方案和进化模型的信息保存在“*.best_scheme.nex”以及“best_scheme_and_models.csv”文件中。
PCGs12RNA数据集
右键单击PCGs12RNA的“Concatenation”结果文件夹,然后选择“Import to ModelFinder”。其他步骤与1.9.1.2-1.9.1.6中所述基本相同(文件夹命名为“PCGs12RNA”),除了在编辑分区的步骤中,使用“Codon Mode (2 sites)”代替“Codon Mode (3 sites)”按钮,具体操作见“PartionFinder2”部分(1.9.2.7)。
AA数据集
右键单击AA的“Concatenation”结果文件夹,然后选择“Import to ModelFinder”。其他步骤与1.9.1.2-1.9.1.6相似(文件夹命名为“AA”),但不需要编辑分区。
如何为外源序列选择最优分区方案和进化模型?
将串联好的(“Fasta”,“Phylip”或“Nexus”格式)序列文件拖到“Alignment File”输入框中,其他步骤与1.9.1.2-1.9.1.5相同,但是需要从头手工编辑“Partition Mode”部分的分区信息。
ModelFinder的其他参数设置详见:http://www.iqtree.org/doc/。
如何使用PartitionFinder2选择最优分区策略和进化模型?
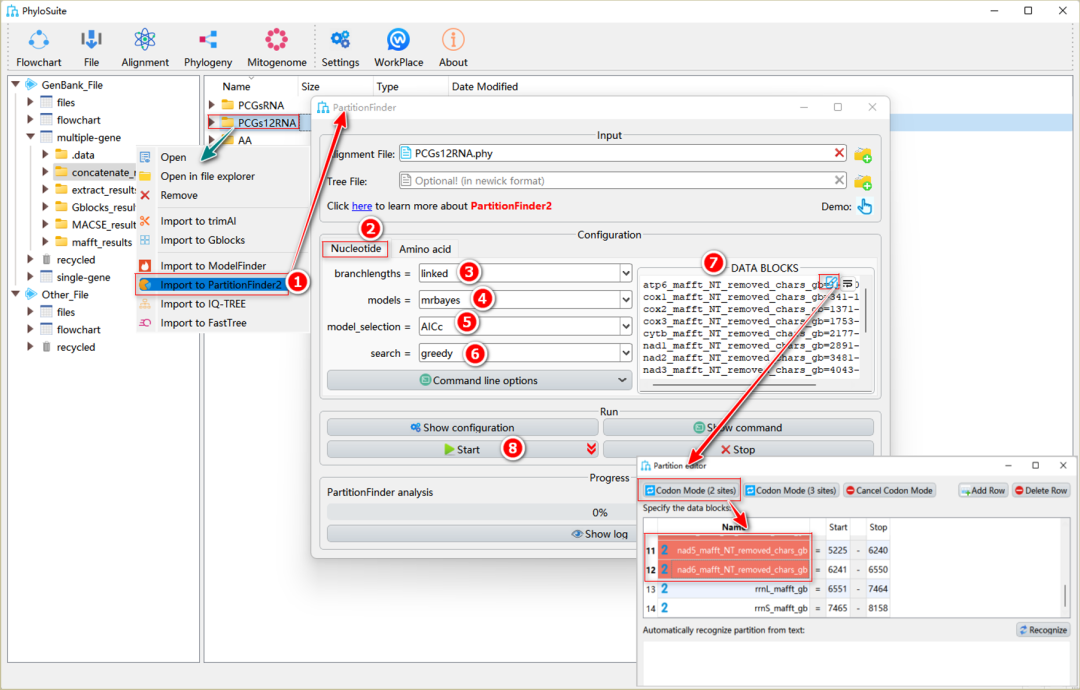
图24. 使用PartitionFinder2进行最优分区策略和进化模型选择
1.9.2.1. 文件输入操作与ModelFinder类似(见1.9.1.1),但选择“Import to PartitionFinder2”。
1.9.2.2. 根据序列类型选择“Nucleotide”或“Amino Acid”标签页。
1.9.2.3. “branchlengths”参数参考ModelFinder(1.9.1.4a)中“Edge-linked”和“Edge-unlinked”的设置。
1.9.2.4. “models”选择适用于下游建树分析的参数。
1.9.2.5. “model_selection”选项对应ModelFinder中的“Criterion”参数,默认设置为PartitionFinder2作者建议的“AICc”。
1.9.2.6. “search”选项默认选择greedy算法,该方法在速度上优于exhaustive search。
1.9.2.7. “DATA BLOCKS”的操作与ModelFinder中“Partition Mode”的“Partition data block edit”相同。
1.9.2.8. 设置结果文件夹名称(在这里命名为“PCGs12RNA”),然后单击“Start”按钮开始运行。
提示:最优模型分区方案和进化模型的信息保存在结果文件夹的“best_scheme_and_models.csv”文件中。PartitionFinder的其他参数设置详见:http://www.robertlanfear.com/partitionfinder/assets/Manual_v2.1.x.pdf。
如何为不同的下游系统发育树重建软件选择最优分区策略和进化模型?
a. IQ-TREE:在ModelFinder的“Model for”下拉框中选择“IQ-TREE”。在PartitionFinder2的“models”下拉框中选择“all”。
b. MrBayes:在ModelFinder的“Model for”选框中选择“MrBayes”。在PartitionFinder2的“models”组合框中选择“mrbayes”。
c. RAxML:在ModelFinder的“Model for”选框中选择“RAxML”。在PartitionFinder2的“Command line options”中选择“--raxml”。
d. BEAST:在ModelFinder的“Model for”选框中选择“BEAST*”。在PartitionFinder2的“models”选框中选择“beast”。
1.10. 重建最大似然法(Maximum Likelihood,ML)系统发育树
什么是基于最大似然法的系统发育分析方法?
likelihood是指基于给定的参数观察数据集得到的概率。对于每个拓扑结构,将通过最大化对数似然(maximizing the log-likelihood)的方法来估计模型中的参数(如分支长度、转换/颠换速率比例等),并使用估计的参数计算出该拓扑结构的log-likelihood(lnL)。最佳的最大似然树是所有可能拓扑结构中lnL最大的树。ML法通常基于比对好的同源序列、进化模型以及多种可能的拓扑结构推导最优系统发育树,推导过程非常耗时,特别是对于大型数据集。例如,当物种数量较多的时候,数据集通常会组合出大量可能的拓扑结构。对于这种问题,目前有两种节省计算时间的方案:1)修剪算法(pruning algorithm),可以减少lnL的重复计算;2)启发式树搜索法(heuristic tree search),例如分支交换算法(branch-swapping algorithms):首先利用随机方法或者是快速建树法(如邻接法或最大简约法)推导出一棵起始树,然后基于该起始树交换部分分支产生多颗备选树,最后根据lnL值来选择最佳树,该方法避免了对所有拓扑结构进行lnL值的计算,进而大量节约分析时间。
为什么使用最大似然法进行系统发育树重建?
与使用简化模型的ML法、简约法以及邻接法相比,基于最优且更符合真实情况的模型(例如,考虑位点间的速率异质性)的ML法受长枝吸引(long-branch attraction,LBA)的影响较小,进而更容易得到接近真实进化历史的拓扑结构。计算机模拟研究表明,当使用碱基/残基的替换速率不恒定、具有高度分化的数据集时,ML法比简约法以及邻接法更稳健(可靠)。此外,ML法还支持多种适用于不同类型数据集的进化模型。
为什么使用IQ-TREE进行系统发育树重建?
在RAxML/ExaML,PhyML,IQ-TREE和FastTree等5种常用的基于ML法的系统发育树重建的软件中,IQ-TREE在多基因串联的数据集中表现最好,得到了最大的似然值。
如何在PhyloSuite中使用IQ-TREE?
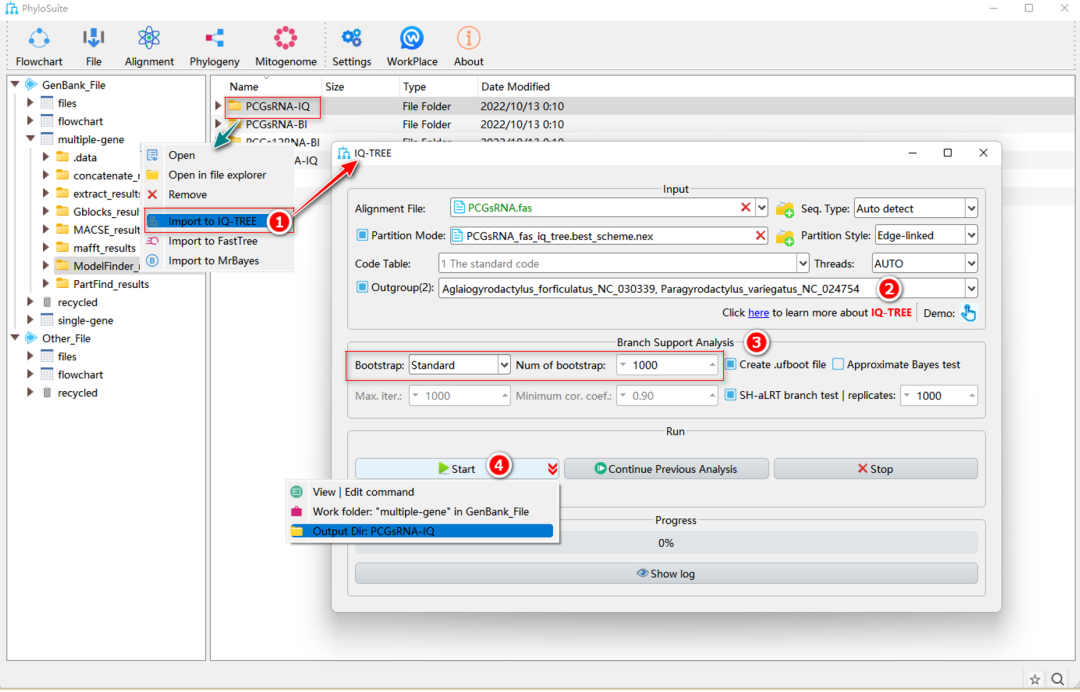
图25. 使用IQ-TREE软件重建ML法系统发育树
1.10.1. 右键单击PCGsRNA数据集的PartitionFinder2或ModelFinder结果文件夹,选择“Import to IQ-TREE”。基因序列将被自动导入到“Alignment File”输入框中;分区结果(最优分区策略和进化模型)将被自动导入“Partition Mode”输入框中。
提示:一旦勾选“Partition Mode”,“Substitution Model Options”参数框中的所有参数将被隐藏。如果未勾选“Partition Mode”,将会根据上游分析得出的最优进化模型在“Substitution Model Options”参数框中自动设置对应参数(见附件5.4最优进化模型选择)。
1.10.2. 选择外群。在系统发育分析中,研究的目标类群称为内群,外群是指一个或多个选定的与内群呈姐妹群关系,或者在进化上略早于内群的物种。远缘的外群可能会影响系统发育树的拓扑结构,例如导致长枝吸引现象。
1.10.3. 设置“Branch Support Analysis”参数框中的参数。Bootstrap是一种评估系统发育树拓扑结构稳定性的算法,值越高拓扑结构越可靠。
注意,如果在“Bootstrap”下拉框中选择参数“Ultrafast”,“Number of Bootstrap”将被自动更改为5000(IQ-TREE手册建议设置1000以上的值);如果选择参数“Standard”,“Number of Bootstrap”将被自动更改为1000。“Ultrafast”模式比“Standard”模式速度更快,但是判定分支可靠性的阈值不同:前者为95,后者为70。
1.10.4. 设置结果文件夹名称(此处命名为“PCGsRNA-IQ”,“PCGs12RNA-IQ”和“AA-IQ”),然后单击“Start”按钮开始运行(图25)。
提示:IQ-TREE的建树结果保存在“*.treefile”文件。
如何使用外源文件重建ML法系统发育树?
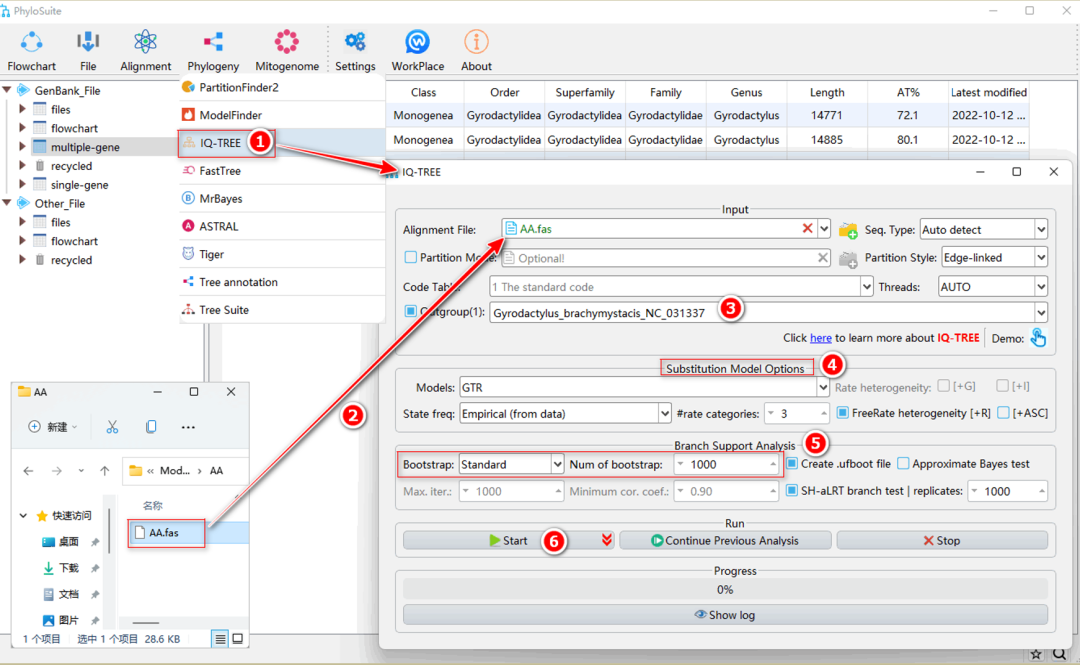
图26. 在IQ-TREE中使用外源文件进行系统发育树重建
将准备好的(“Fasta”、“Phylip”或“Nexus”格式)文件拖到“Alignment File”输入框中;分区文件拖到“Partition Mode”输入框中并勾选“Partition Mode”(可选)。如果没有分区文件,在“Substitution Model Options”参数框中为数据设置最适合的模型参数(图26)。其他步骤与1.10.2-1.10.4中所述的相同。IQ-TREE分析的其他参数设置详见:http://www.iqtree.org/doc/。
1.11. 重建贝叶斯法(Bayesian inference,BI)系统发育树
什么是基于BI法的系统发育分析?
BI在系统发育中是指贝叶斯推断(Bayesian Inference)。BI法将后验概率值(基于数据、先验概率和替代模型计算得到)最高的树作为最优树。历史上,BI法因对计算资源需求过大而发展缓慢,但随着马尔可夫链蒙特卡洛(MCMC)算法的发展,BI法也慢慢流行起来。
为什么使用BI法进行系统发育树重建?
BI法是处理进化生物学复杂问题的强大算法,适用于大数据集的系统发育树重建、自然选择检测以及最优进化模型选择。自从Huelsenbeck等人发布MrBayes软件以来,BI法就成为了系统发育分析的流行算法。另外,进化模型的发展也进一步促进了BI法的普及。因此,研究人员随后开发出了超过十款基于BI的软件程序(见引文参考文献[57]的表1)。
如何在PhyloSuite中重建BI法系统发育树?
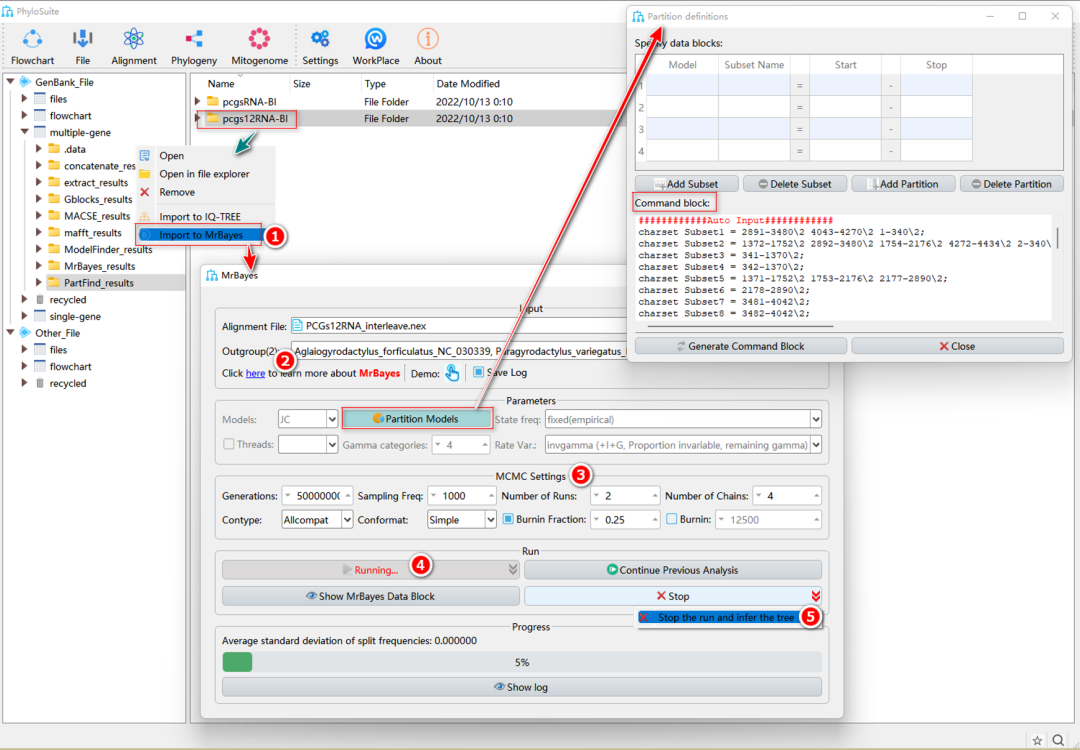
图27. 在PhyloSuite中使用MrBayes进行系统发育树重建
1.11.1. 右键单击“PCGsRNA”、“PCGs12RNA”或“AA”的PartitionFinder2或ModelFinder的结果文件夹,然后选择“Import to MrBayes”。基因序列文件将被导入“Alignment File”输入框,最优分区策略和进化模型将被导入“Partition Models”,双击该按钮可以查看分区结果(注意,如果此按钮选中,Models”等参数的设置将会失效,即基于分区结果建树)。
1.11.2. 选择外群。
1.11.3. “MCMC Settings”参数设置。
a. Generations:指定单次运行的MCMC世代数。建议设置一个较大的代数,因为当结果收敛时,可以随时在PhyloSuite中结束运行并推导系统发育树。
b. Sampling Freq:定义马尔可夫链被采样的频率(即每多少代采样一次),该值和MCMC的世代数决定产生的样本的数量。如果设置的值过小,输出的结果将会占较大内存。PhyloSuite(v1.2.3)使用1000作为默认值,即运行1,000,000代将产生1000个采样统计结果(每个结果包含一棵树)。
c. Nrun:同时运行的独立分析数量,默认值为2。
d. Nchains:同时运行的马尔可夫链数。默认值为4,包括3条“热链”和1条“冷链”(详见MrBayes手册)。
f. Contype:一致树的类型。“Halfcompat”:生成50%多数规则树(majority-rule tree),即后验概率值(BPP)小于0.5的进化枝将被视为多歧枝。“Allcompat”,BPP小于0.5的进化枝依然视为二歧枝,该参数与PAUP中勾选了“Show frequencies of all observed bipartitions”选项的50%多数规则树相似。PhyloSuite(v1.2.3)将以“Allcompat”为默认参数。
g. Conformat:一致树的格式。“Simple”:生成简单的一致树,可以被多数下游程序(例如iTOL)识别。“Figtree”:结果文件中包含丰富的可以被Figtree软件识别的统计信息,但iTOL等软件无法识别。PhyloSuite(v1.2.3)默认使用“Simple”。
h. Burnin Fraction:在BI分析中,马尔可夫链刚开始通常会经历一段不稳定状态,因此为了参数估计更可靠,同时减少模拟误差(simulation errors),通常会选择删除早期不稳定的马尔可夫链运行结果,这一过程被称为“burnin”。“Burnin Fraction”值默认为0.25,即前25%样本被删除。此外,“burnin”选项可以直接指定要删除的样本数。
1.11.4. 设置结果文件夹名称(在这里命名为“PCGsRNA-BI”,“PCGs12RNA-BI”和“AA-BI”),然后单击“Start”按钮开始运行。
1.11.5. BI运行收敛后,点击“Stop”按钮右侧的下拉菜单,选择“Stop the run and infer the tree”,得到树和统计结果文件(图27)。
如何评估BI运行是否收敛?
MrBayes使用MCMC进行系统发育的贝叶斯推断,MCMC运行的理想状态是达到目标后验概率分布。例如,2个运行(run)生成的树样本应该非常相似。当MrBayes开始运行时,“Progress”框将实时显示“Average standard deviation of split frequencies”(ASDSF)值。一般来说,当ASDSF值低于0.01时,便可以认为BI运行收敛,此时可以停止运行获得结果文件(具体操作见1.11.5)。
此外,还有2种指标可用于评估MCMC运行是否收敛。第一种是potential scale reduction factor(PSRF)指标,当运行收敛时,所有参数的PSRF应接接近1.0。另一个是effective sample size(ESS),其值高于100才算收敛。这两个指标都可以在log文件中找到。(详见MrBayes说明书)
如果BI运行结束但不收敛怎么办?
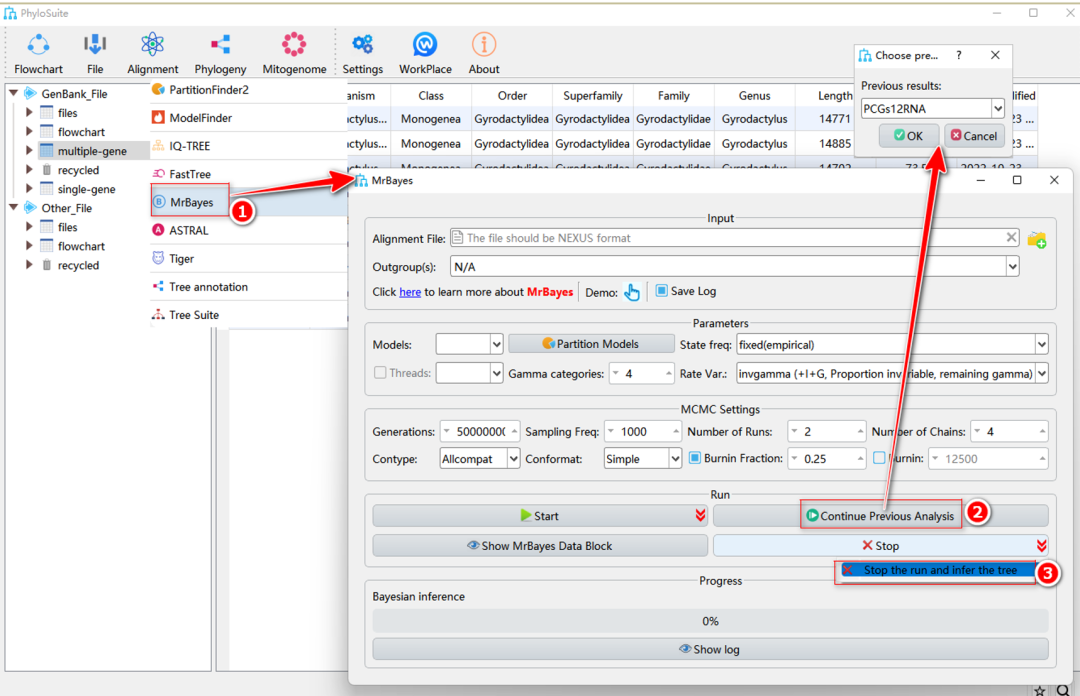
图28. MrBayes续跑
单击“Continue Previous Analysis”按钮,当前工作文件夹(“multiple-gene”)中的所有MrBayes的结果(如“PCGs12RNA”)将在选框中弹出,选择未收敛的结果,然后单击“OK”继续运行(图28)。
如何使用外源序列重建BI系统发育树?
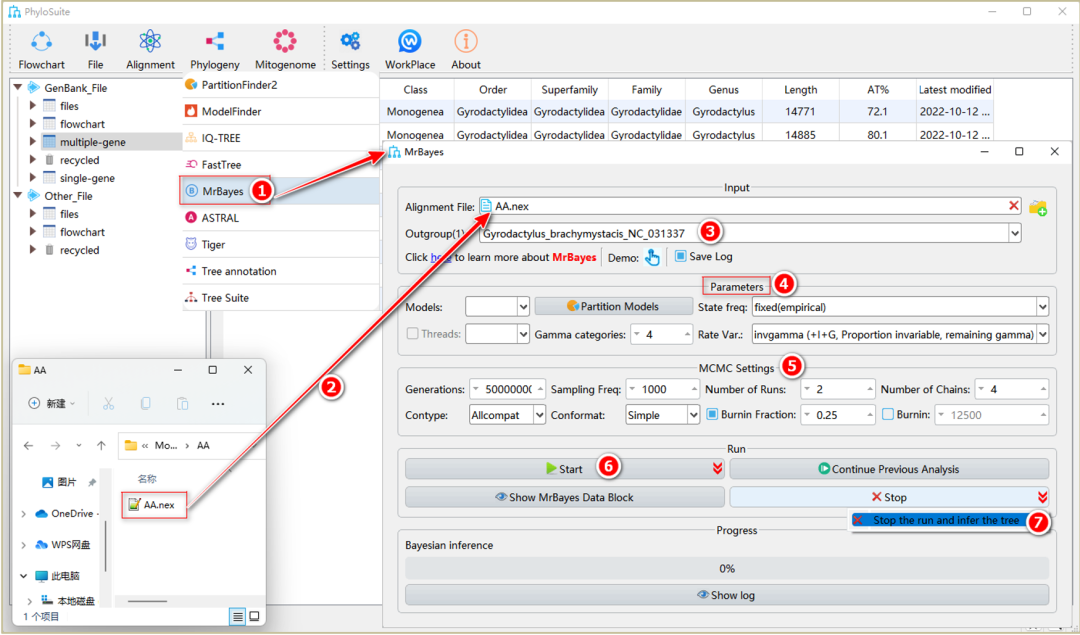
图29. 使用MrBayes和外源序列文件进行系统发育树重建
将准备好的(“Nexus”格式)文件拖到相应的输入框中,然后在PhyloSuite中设置参数,例如最优进化模型,其他步骤与1.11.2-1.11.5类似(图29)。
MrBayes的其他参数设置详见:http://mrbayes.sourceforge.net/manual.php。
提示:BI树结果保存在“*.con.tre”文件中。
2.基于系统发育树的统计分析
相关分析均在PhyloSuite中“Phylogeny”菜单下的“TreeSuite”功能中。
2.1. TreeSuite的功能介绍
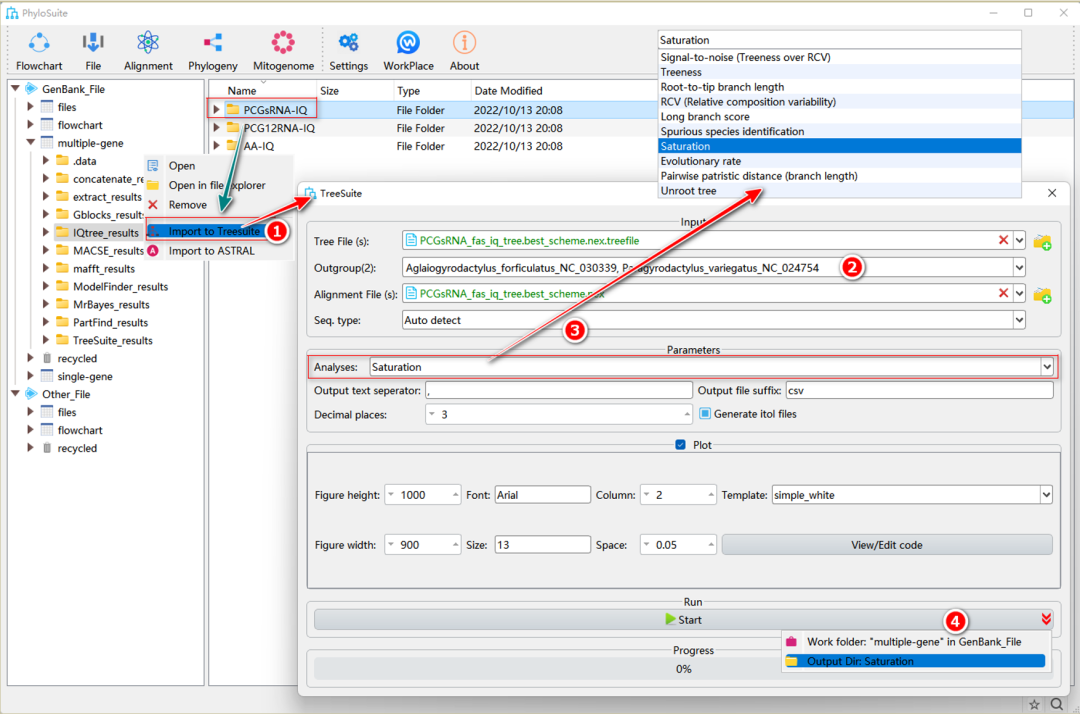
图30. TreeSuite的操作
2.1.1. 右键单击“IQ-TREE”的结果文件夹,然后选择“Import to TreeSuite”。相应的树文件和序列文件将被分别导入相应的“Tree File (s)”和“Alignment File (s)”输入框中。
提示:“Tree File (s)”输入框仅接受标准的“Newick”、“Nexus”、“Nexml”和“Phyloxml”格式;“Alignment File (s)”输入框仅接受标准的“Fasta”、“Nexus”和“Phylip”格式。此外,我们可以同时导入多个树和MSA文件。如果树文件与对应的MSA文件名称相同,则将它们进行组合分析;否则,PhyloSuite将先对树文件和MSA文件进行两两自由组合,然后开始分析。
2.1.2. 选择外群。注意,当导入多个树文件时,只有第一个树文件中的物种可以选择外群;因此要确保所有树文件具有相同的外群。
2.1.3. 选择所需的分析(详细信息见下文)。
2.1.4. 参数配置完成后,设置结果文件夹名称,然后单击“Start”按钮开始分析(图30)。
提示:分析结果可在“TreeSuite”的结果文件夹中查看。“Signal-to-noise(Treeness over RCV)”和“Saturation”分析同时需要MSA文件和树文件;“RCV(Relative composition variability)”分析仅需要MSA文件;其他分析(例如“root-to-tip branch length”)只需要树文件。
2.2. 替换饱和分析
什么是替换饱和?
替换饱和是指MSA中的某一位点发生了多次碱基替换,因此导致观察到的替换数小于真实替换数。替换饱和度可以根据序列的真实替换数与观察到的替换数的比值来推断。
以观察到的替换数(pairwise distance)为因变量,实际替换数(patristic distance)为自变量,进行线性回归拟合后,R平方(r2)可以反映回归模型中自变量可以解释的因变量变异的百分比,即反映替换饱和度的高低。如果某一位点发生了多次替换,则因变量将小于自变量,导致r2和回归线斜率值较低。
为什么要分析替换饱和?
MSA中的替换饱和位点可能会产生“噪音”,从而影响真实系统发育关系的推导。替换饱和现象通常在包含远缘物种或快速进化的数据集中非常明显,识别和去除存在替换饱和的位点可以提高系统发育分析的可靠性(详见引文参考文献 [72,73])。
如何在PhyloSuite中进行替换饱和度分析?
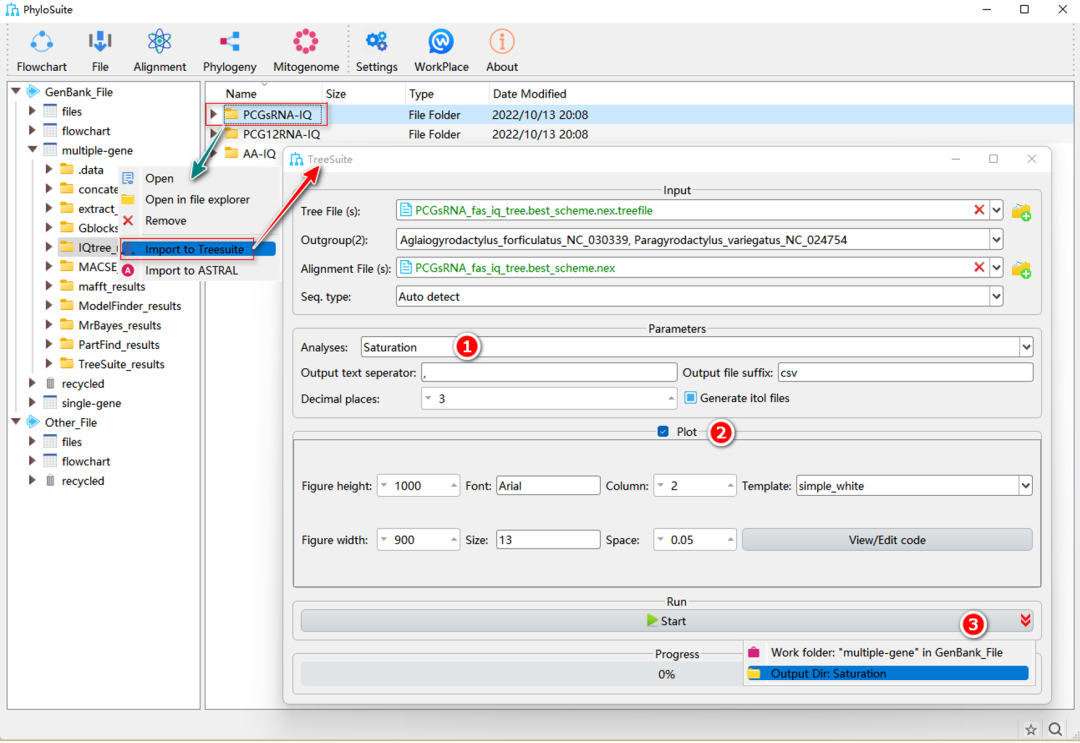
图31. TreeSuite中的饱和度分析
2.2.1. 选择“Saturation”分析。
2.2.2. 勾选“Plot”选框,绘制期望距离(x)和观察距离(y)的回归图,用于评估饱和程度。
2.2.3. 设置结果文件夹名称(命名为“Saturation”),然后单击“Start”按钮开始进行饱和度分析(图31)。
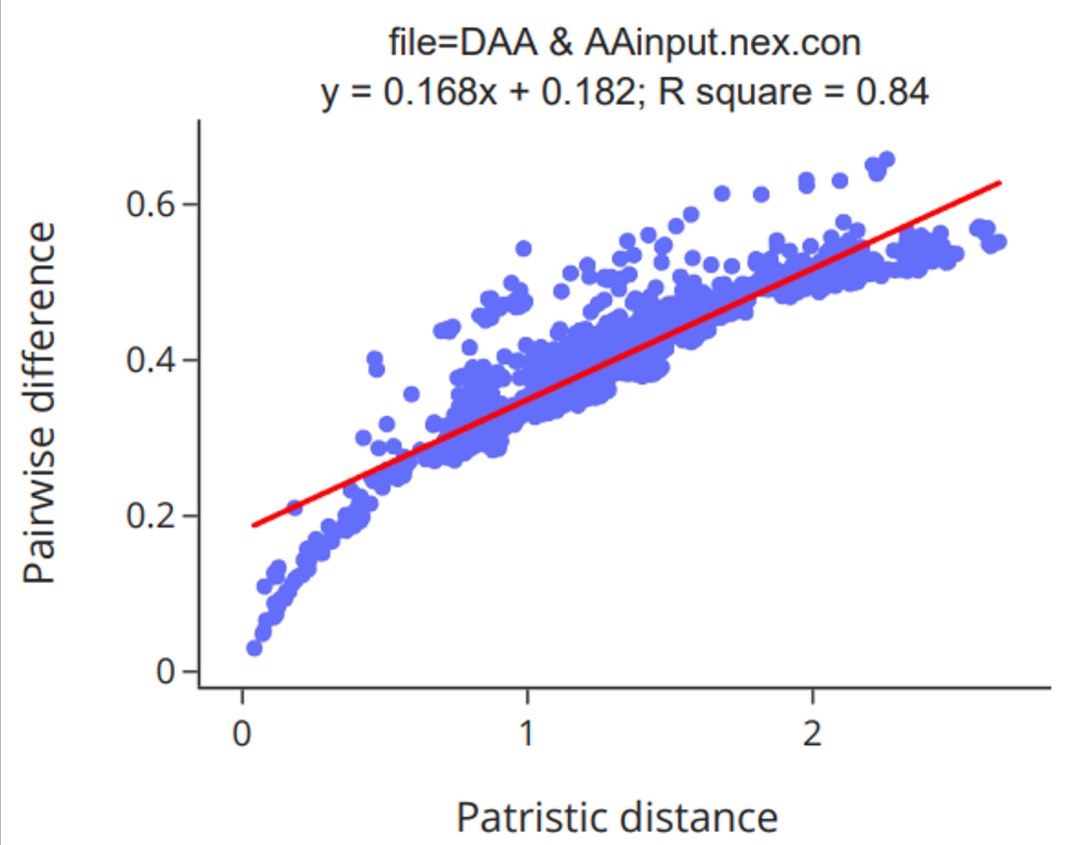
图32. 饱和度的回归分析图
图32展示了饱和度回归分析图,R2值越大,斜率越接近于1,代表饱和程度越低。在结果文件夹中,“saturation.regression.pdf”和“saturation.regression.html”两个文件是回归分析图;“saturation.regression.csv”文件包含回归分析的详细统计信息;“saturation.csv”文件包含成对物种的观察距离pairwise difference和期望距离patriscic distance信息;“plot_data.tsv”文件包含用于绘图的数据;“plot_data.cmd.py”是用于绘图的python脚本。
2.3 长枝分数(long-branch score)
什么是长枝分数?
长枝分数可衡量每个物种对长枝吸引现象(long-branch attraction artefacts,LBA)的贡献,计算公式如下:
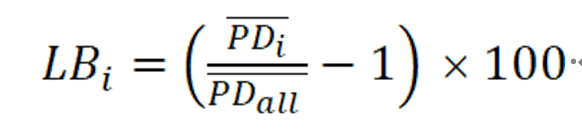

为什么计算长枝分数?
长枝分数的计算可用于在系统发育分析中识别并排除有可能产生LBA的物种。与另一种衡量物种LBA的指标root-to-tip距离相比,长枝分数不会随着根(祖先节点)的变化而变化。
如何在PhyloSuite中计算长枝分数?
图33. TreeSuite中长枝分数的计算
2.3.1. 选择“Long branch score”分析。
2.3.2. 设置结果文件夹名称(在这里命名为“Long-branch-score”)后单击“Start”按钮开始计算长枝分数(图33)。
Tips:在结果文件中,“Long branch scores.csv”包含每个物种的长枝分数;“Long branch scores overall.csv”包含整个树(所有物种)的长枝分数;“* itol.txt”文件可用于在iTOL中绘制条形图。
2.4. 鉴定“噪音物种(spurious species)”
什么是噪音物种?
噪音物种一般指在数据集中表现出特殊的进化模式并且可能会导致LBA的物种。噪音物种鉴定的阈值一般是20,即终末枝的枝长至少比整个树中所有分支长度(包括内部节点和终末节点)的平均值大20倍。该阈值可根据不同情况自定义。
为什么要鉴定噪音物种?
去除噪音物种可以稳定系统发育树的拓扑结构。
如何在PhyloSuite中鉴定噪音物种?
图34. TreeSuite中噪音物种的鉴定
2.4.1. 在“Analyses”下拉框中选择“Spurious species identification”。
2.4.2. 设置噪音物种识别的阈值(默认为20)。
2.4.3. 参数配置完成后,设置结果文件夹名称(这里命名为“Spurious-species”)后单击“Start”按钮开始运行(图34)。
提示:在结果文件夹中,“Spurious species.csv”文件包括鉴定出的噪音物种、它们的枝长和所有物种及内部节点枝长的中位数。
2.5. Treeness、RCV和信噪比
什么是Treeness?
Treeness是一种可以用于判断系统发育分析中信噪比大小的指标,通过进化树内部分支长度的总和除以所有分支长度的总和来计算。在进化树中,内部分支长度代表物种之间的共有衍征和祖征(物种从共同祖先遗传下来的特征)的积累;终末节点枝长代表每个物种的独有衍征的累积。
什么是RCV?
RCV(relative composition variability)是衡量MSA中物种的平均组成变异性的指标。计算公式如下:
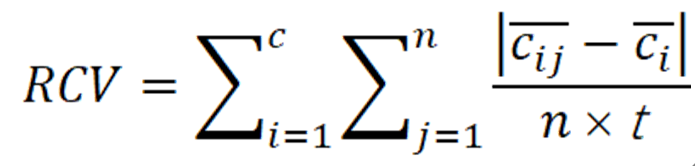

信噪比是什么?
信噪比是指系统发育信号(用于推导“真实”进化树的信号)与数据噪声(可能影响 “真实”进化树推导的信号,例如异质性)之间的比值,通过Treeness除以RCV来计算。
为什么要计算Treeness,RCV和信噪比?
Treeness可用于评估系统发育信号量。例如,如果姐妹分支共享一个非常短的祖先节点枝长,表明两者之间存在的祖征可能相对较少,从而导致它们在系统发育关系推导过程中难以被解析为姐妹分支。因此,低Treeness很容易造成基于简约法、邻接法和使用简化模型的ML法的系统发育树产生错误拓扑结构。
RCV可用于评估MSA中序列的碱基组成差异程度,也称为数据的异质性或“噪声”。
Treeness/RCV称为信噪比。具有高信噪比的数据集有更大概率重建出稳定的系统发育树。
如何在PhyloSuite中计算Treeness、RCV和信噪比?
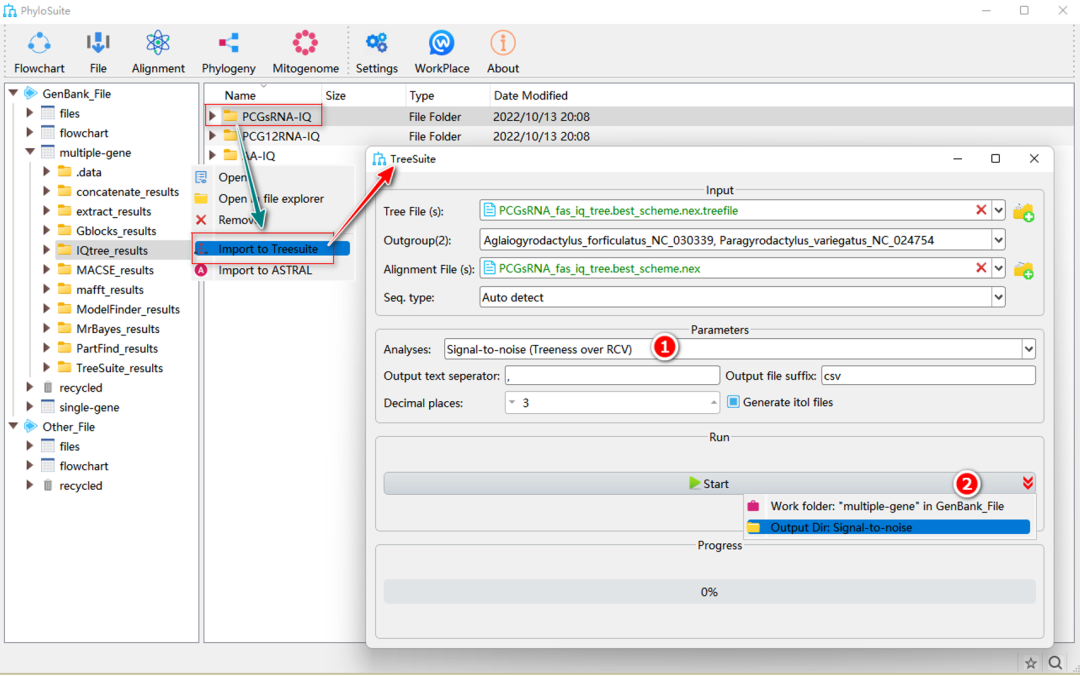
图35. Treeness、RCV和信噪比的计算
2.5.1. 选择“signal-to-noise(Treeness over RCV)”、“Treeness”或“RCV(relative composition variability)”分析。
2.5.2. 参数配置完成后,设置结果文件夹名称(这里命名为“Signal-to-noise”)后单击“Start”按钮开始计算(图35)。
提示:在“signal-to-noise”分析的结果文件夹中,“signal-to-ratio.csv”文件包括所有物种的信噪比、Treeness和RCV的值。在“Treeness”的结果文件夹中,“treeness.csv”文件包括所有物种的Treeness值。在“RCV(relative composition variability)”的结果中,“Species.RCV.csv”包括每个物种的RCV值,“RCV.csv”包括所有物种的RCV值。
2.6 两种类型的分支长度计算(Pairwise patristic distance和root-to-tip branch length)
Pairwise patristic distance和root-to-tip branch length是什么?
系统发育树的树枝代表遗传信息从祖先到后代的传递路径,即代表了祖先节点到后裔节点之间的遗传变异量。
Pairwise patristic distance代表连接系统发育树中两个终末节点(通常为两个物种)的分支长度之和。
root-to-tip branch length:从根节点到终末节点的分支长度。
为什么要计算Pairwise patristic distance 和root-to-tip branch length?
Patristic distance可用于估计进化树中物种对之间的遗传距离,而root-to-tip branch length可用于代表物种或基因的碱基替换率(进化速率)。由于系统发育树中所有物种与根节点之间的时间跨度相同,因此,如果所有物种的碱基替换率相同,则系统发育树中所有物种都应有相同的root-to-tip branch length,root-to-tip branch length不同代表数据集中物种之间的进化速率不同。
如何在PhyloSuite中计算Pairwise patristic distance 和root-to-tip branch length?
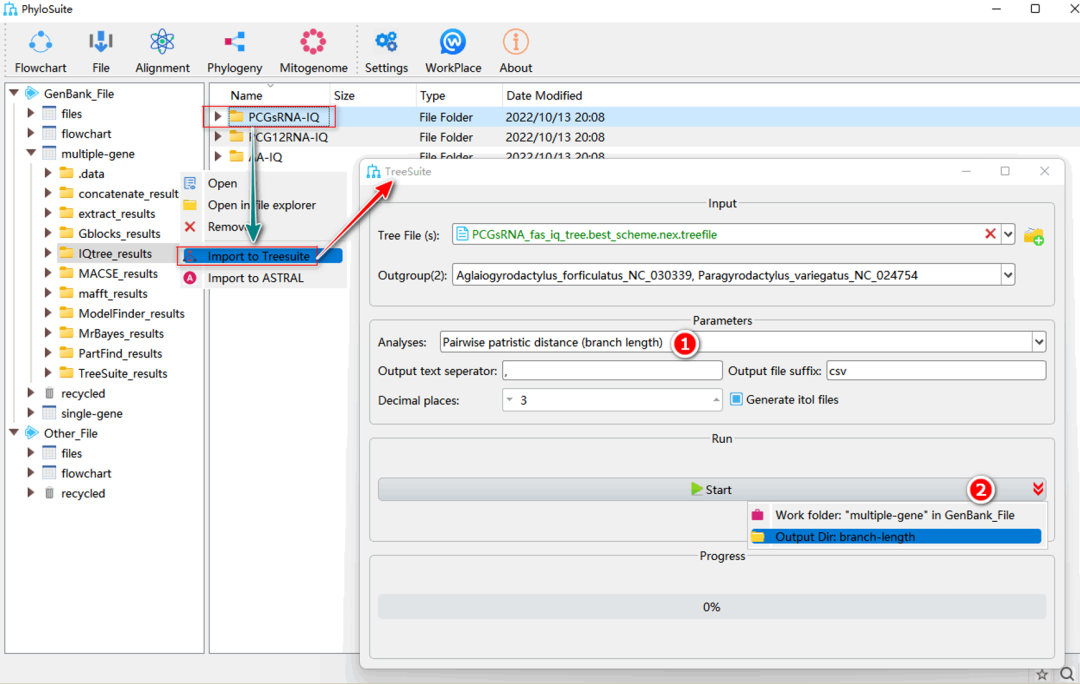
图36. TreeSuite中Pairwise patristic distance和root-to-tip branch length的计算
2.6.1. 选择“Pairwise patristic distance(branch length)”或“root-to-tip branch length”分析。
2.6.2. 参数配置完成后,设置结果文件夹名称(这里命名为“branch-length”)后单击 “Start”按钮开始计算(图36)。
在Pairwise patristic distance的结果文件夹中,“Patristic distance.csv”文件包含成对物种之间的距离值,“*matrix.csv”文件包含距离矩阵。在root-to-tip计算的结果文件夹中,“root-to-tip-branch-length.csv”包含每个物种的root-to-tip值。“* .itol.txt”文件可用于在iTOL中绘制root-to-tip的条形图。
2.7. 进化率
进化率是什么?
进化率是指基因或物种在一定时期内的遗传改变的速率。在PhyloSuite中,进化率可以通过将所有分支(内部分支和终末枝)的长度总和除以终末节点的总数来计算。
为什么计算进化率?
蛋白质、核苷酸等分子标记的进化速率在进化生物学中非常重要。例如,可以通过单基因树估算每个基因的进化速率,进而识别缓慢进化和快速进化的基因。
如何在PhyloSuite中计算进化速率?
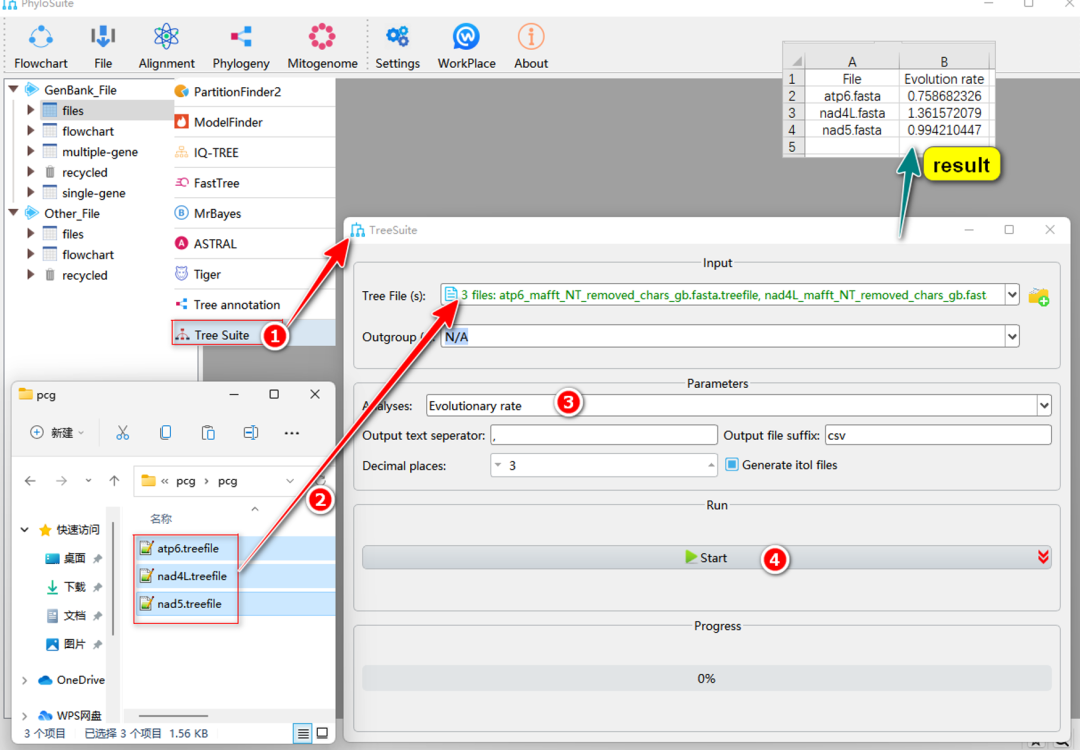
图37. TreeSuite中计算进化速率
2.7.1. 选择“Evolution rate”。
2.7.2. 将准备好的树文件(例如IQ-TREE的“*.treefile”)拖到“Tree File (s)”输入框中。参数配置完成后,设置结果文件夹名称(此处命名为“Evolution-rate”)后单击“Start”按钮开始运行(图37)。
Tips:在“Evolution rate”的结果文件夹中,“Evolution rate.csv”文件包含进化速率的计算结果。
2.8. “有根树”转“无根树”
什么是无根树?
有根树是指具有一个祖先节点(称为根)的系统发育树,该节点在进化上早于系统发育树中的所有其它节点,代表了系统发育树中所有现存物种的共同祖先。
无根树是没有确定共同祖先节点的系统发育树,因此它没有时间方向,仅展示现存物种之间的进化关系。
如何在PhyloSuite中将有根树转换为无根树?
在PhyloSuite中,基于ETE3的“Unroot tree”分析可将有根树转换为无根树。
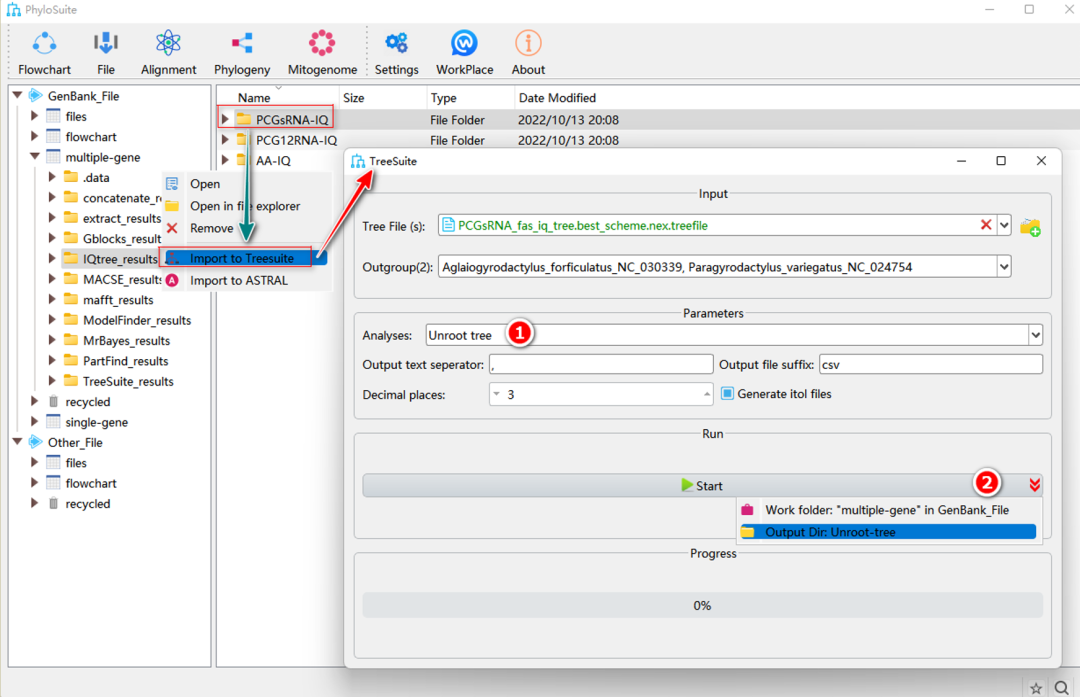
图38. 在TreeSuite中将有根树转化为无根树
2.8.1. 选择“Unroot tree”。
2.8.2. 参数配置完成后,设置结果文件夹名称(这里命名为“Unroot-tree”)后单击“Start”按钮开始运行(图38)。
提示:转换后的无根树保存在(*.nwk)文件中。
2.9. 多歧枝处理
什么是多歧枝?
多歧枝是指系统进化树中具有三个或更多直接后代的内部节点的分支,只有两个直接后代的内部节点的分支称为二歧枝。
为什么要解决多歧枝?
当物种之间的进化关系不确定或没有足够的信息来推导系统发育关系时,可能会产生多歧枝。由于某些下游分析软件不支持多歧枝,因此需要将其转换为二歧枝。
如何在PhyloSuite中解决多歧枝?
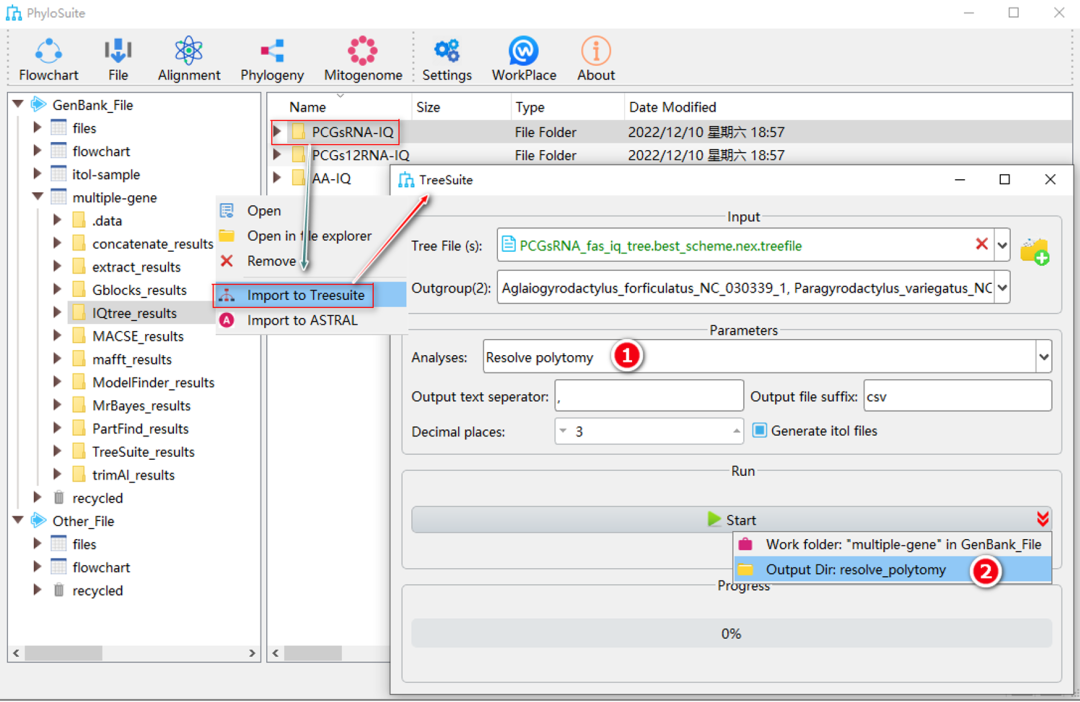
图39. 在TreeSuite中处理多歧枝
2.9.1. 选择“Resolve polytomy”。
2.9.2. 参数配置完成后,设置结果文件夹名称(在这里命名为“resolve_polytomy”),然后单击“Start”按钮将多歧枝转化为二歧枝(图39)。
提示:转换后的二歧树保存在(*.nwk)文件中。
3.NOTES
在NCBI的分类数据库(https://www.ncbi.nlm.nih.gov/taxonomy)中查询物种对应的密码表(图40)。
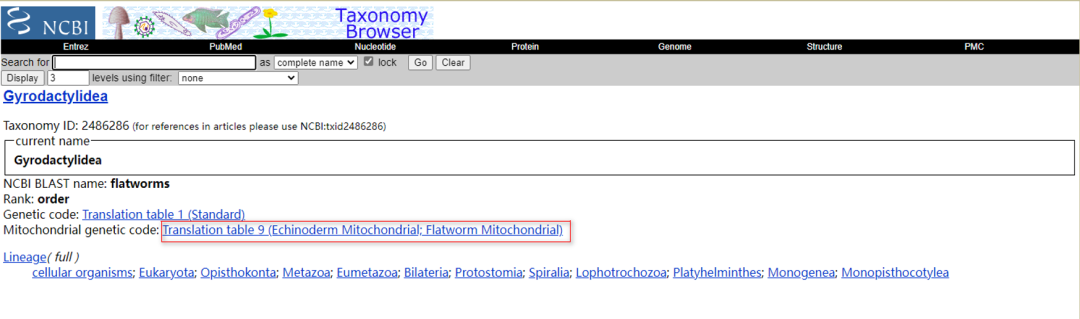
图40. Gyrodactylidea的线粒体基因组对应的遗传密码
4.故障排除(troubleshooting)
表1总结了一些常见问题的故障排除建议,有关其他问题和说明详见附件第8节(故障排除)。
表1. 故障排除:PhyloSuite用户遇到的一些常见问题

附 件
5.单基因系统发育分析
与“多基因联合系统发育分析”部分重复的内容不再详述。
5.1. 根据GenBank IDs下载序列
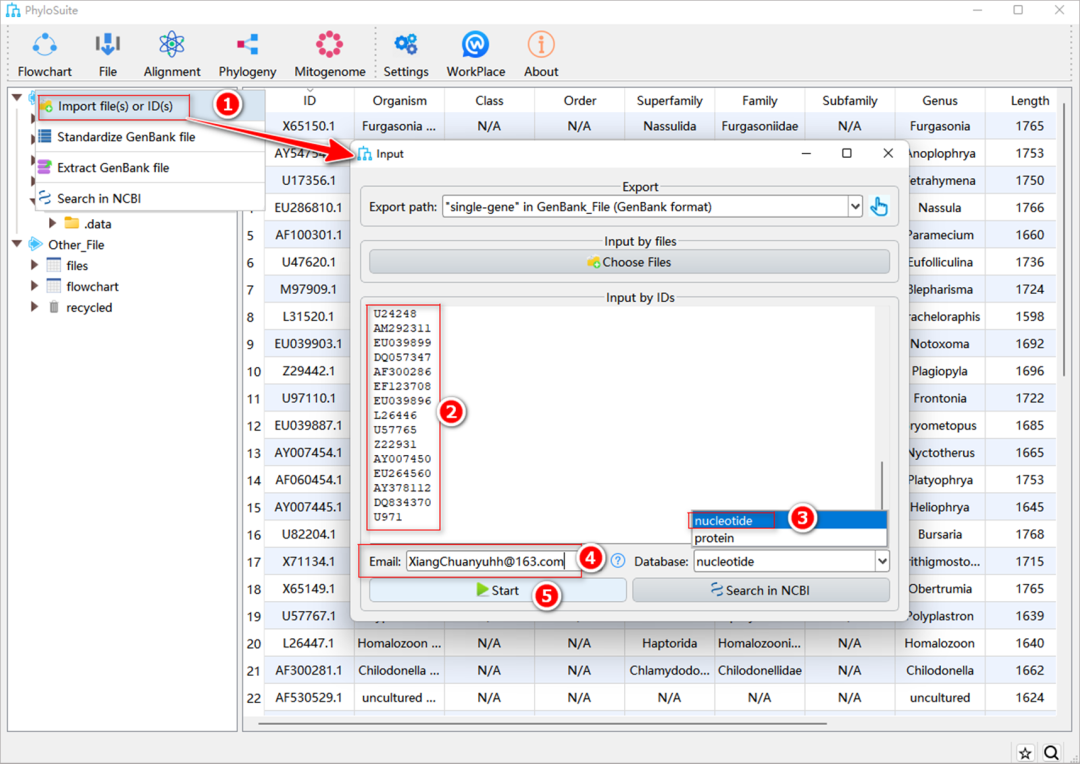
图S1. 通过IDs下载序列
5.1.1. 创建一个“single-gene”工作文件夹并打开。单击“File”菜单栏,然后选择“Import file (s) or ID (s)”打开窗口。
5.1.2. 将所有准备好的序列ID复制到“Input by IDs”输入框中(空格,换行符,制表符等都可作为分隔符)。
5.1.3. 选择与序列匹配的数据库。
5.1.4. 输入电子邮箱(告诉NCBI谁在下载序列)。
5.1.5. 单击“Start”下载序列(图S1)。
5.2. 单基因序列的提取
过滤序列和获取分类信息操作与“1.3. 删除冗余序列”的步骤相同。
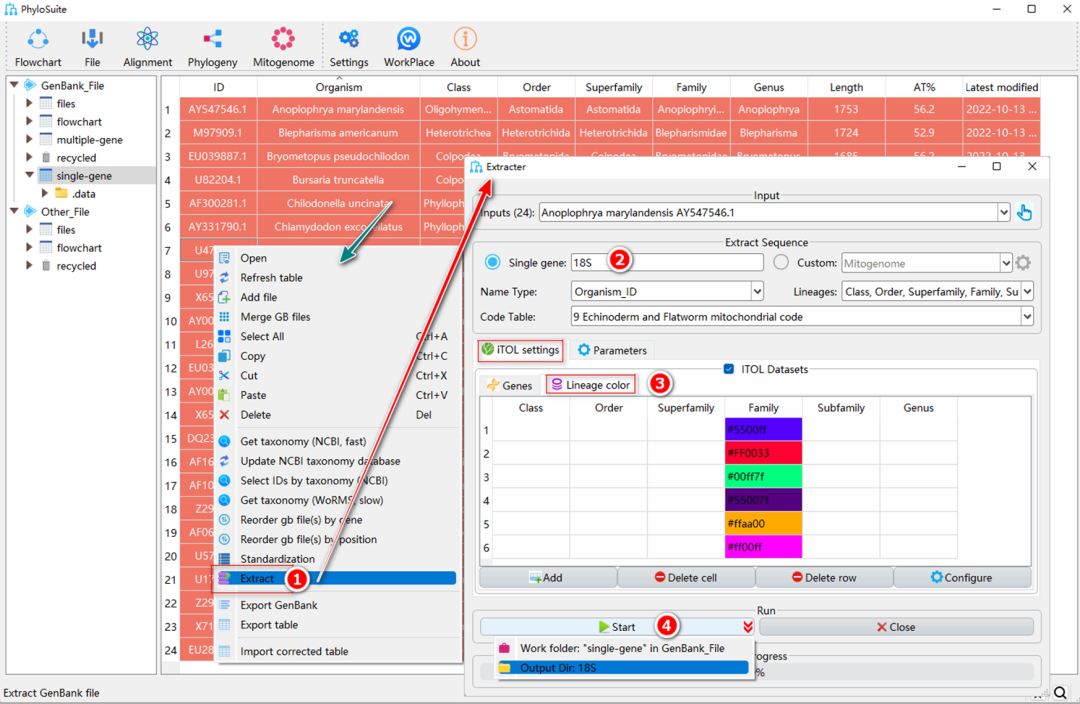
图S2. 单基因序列提取
5.2.1. 按Ctrl+A全选导入的序列,单击右键打开菜单后选择“Extract”。
5.2.2. 在“Extracter”窗口中,选中“Single gene”,然后在输入框中输入基因名称(例如“18S”)。
5.2.3. “Single gene”模式仅需设置“iTOL settings”中的“Lineage color”。
5.2.4. 设置输出文件夹名称(此处设置为“18S”),然后单击“Start”按钮开始提取(图S2)。
提示:在“Single gene”模式下无需设置密码表。
5.3. 序列比对
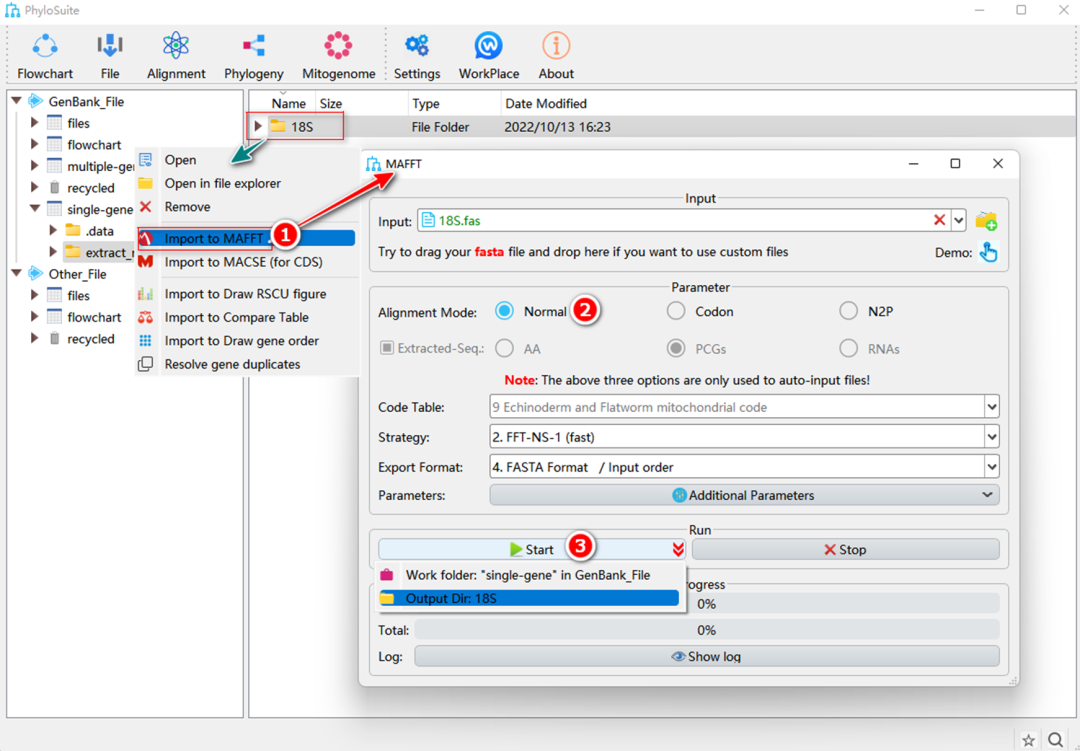
图S3. 单基因序列比对
5.3.1. 右键单击18S的“Extract”结果文件夹,然后选择“Import to MAFFT”。
5.3.2. “Alignment mode”选择“Normal”。注意,“Single gene”模式下“Extracted-Seq”选框不可用。
5.3.3. 设置输出文件夹名称,然后单击“Start”按钮开始比对(图S3)。提示:由于18S不是蛋白编码基因,因此不需要在MACSE进行比对优化。
5.4. 最优进化模型选择
什么是模型选择?
在分子系统发生学中,进化替代模型是一种描述替代模式和速率的马尔可夫链模型。最优进化模型选择是指从大量候选的进化模型中选择最能反映序列替换过程的模型。
为什么选择最优进化模型?
最优进化模型可用于在最大似然法中计算似然值,对正确识别位点的多重替换现象以及描述位点之间和不同进化谱系内的替代模式至关重要。由于不同的基因和物种的进化速率通常不同,错误的模型可能会降低系统发育树重建的准确性。但有证据表明,直接选择参数最丰富的模型(如GTR)作为最优进化模型同样能获得可靠的系统发育拓扑结构。
为什么要使用ModelFinder?
ModelFinder在模型与数据的拟合程度上往往优于其他模型选择软件,除此之外,速度优势(速度上比大多数模型选择方法,例如jModelTest和ProtTest快10到100倍)也使得ModelFinder在大型数据集中更适用。
如何在PhyloSuite中选择最优进化模型?
单基因序列不需要修剪、串联和分区,因此序列比对完成后即可进行最优模型选择。
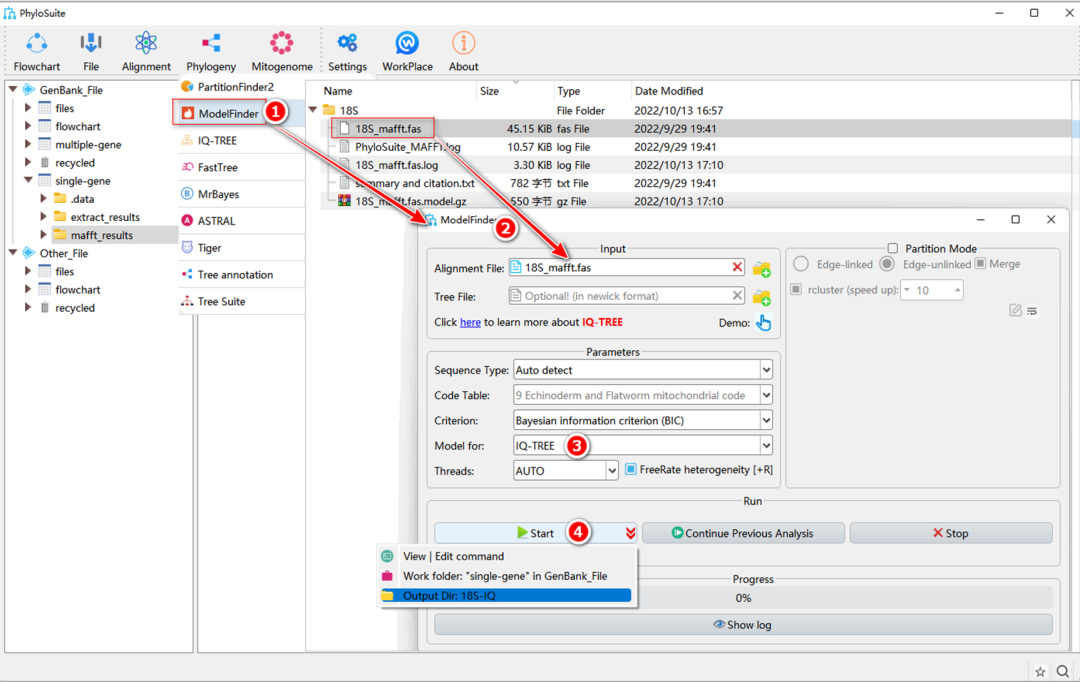
图S4. 使用ModelFinder进行最优进化模型选择
5.4.1. 在“Phylogeny”菜单中选择“ModelFinder”。
5.4.2. 在MAFFT的结果文件夹中找到“*_mafft.fas”文件后,将其拖到“Alignment file”输入框中。
5.4.3. 在“Model for”下拉框中,选择用于系统发育树重建的下游软件。
5.4.4. 参数配置完成后,设置结果文件夹名称(在这里其命名为“18S-IQ”),然后单击“Start”按钮开始运行(图S4)。
提示:基于三个标准(AIC,BIC、AICc)的最优进化模型选择结果可在“*iq_tree.iqtree”文件中查看。
如何为MrBayes选择最优进化模型?
操作与5.4.1-5.4.4中相同,但在“Model for”组合框中选择“MrBayes”。基于三个标准(AIC、BIC和AICc)的最优进化模型可以在“*mrbayes.iqtree”文件中找到。
5.5. 使用IQ-TREE进行最大似然树重建
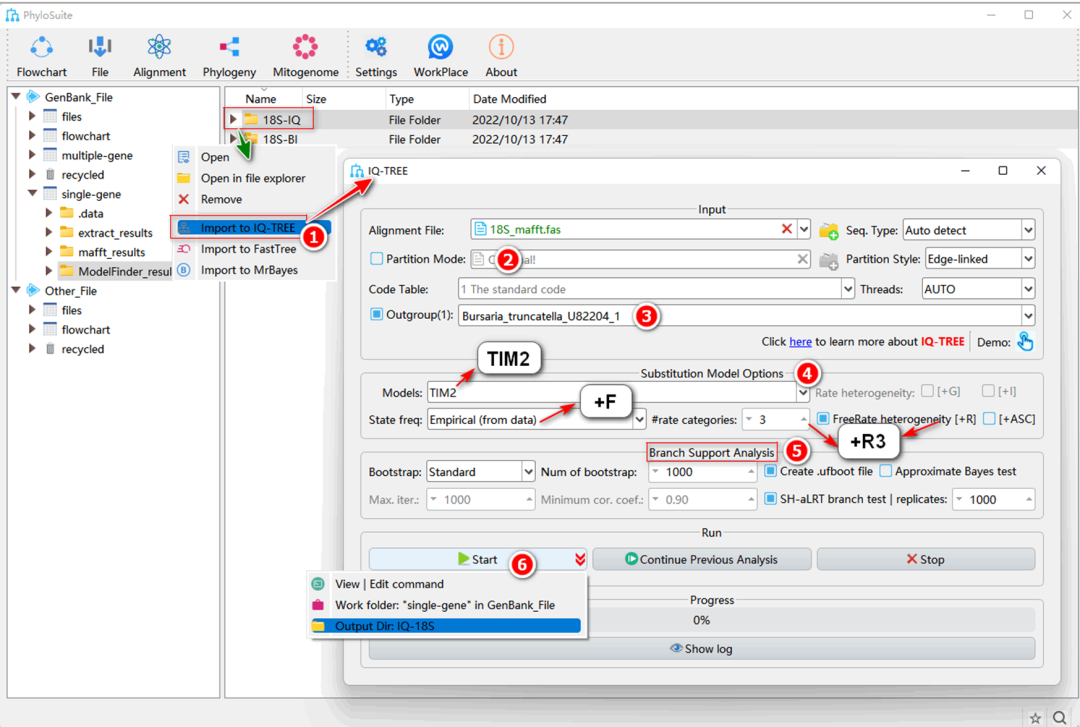
图S5. 使用IQ-TREE进行最大似然树重建
5.5.1. 右键单击“ModelFinder”的结果文件夹,然后选择“Import to IQ-TREE”。MSA文件将被自动导入“Alignment File”输入框中,最优进化模型(TIM2+F+R3)将在“Substitution Model Options”中自动设置。
5.5.2. 确认“Partition Mode”取消选中。
5.5.3. 选择外群。
5.5.4. 最优进化模型参数已在导入时自动设置。
5.5.5. 根据IQ-TREE手册,单基因推荐选择“Bootstrap”下拉框中的“Standard”参数,并将“Num of bootstrap”设置为1000。
5.5.6. 参数配置完成后,设置结果文件夹名称(在这里其命名为“IQ-18S”),然后单击 “Start”按钮开始重建系统发育树(图S5)。
提示:构建好的系统发育树保存在“*.treefile”文件中。
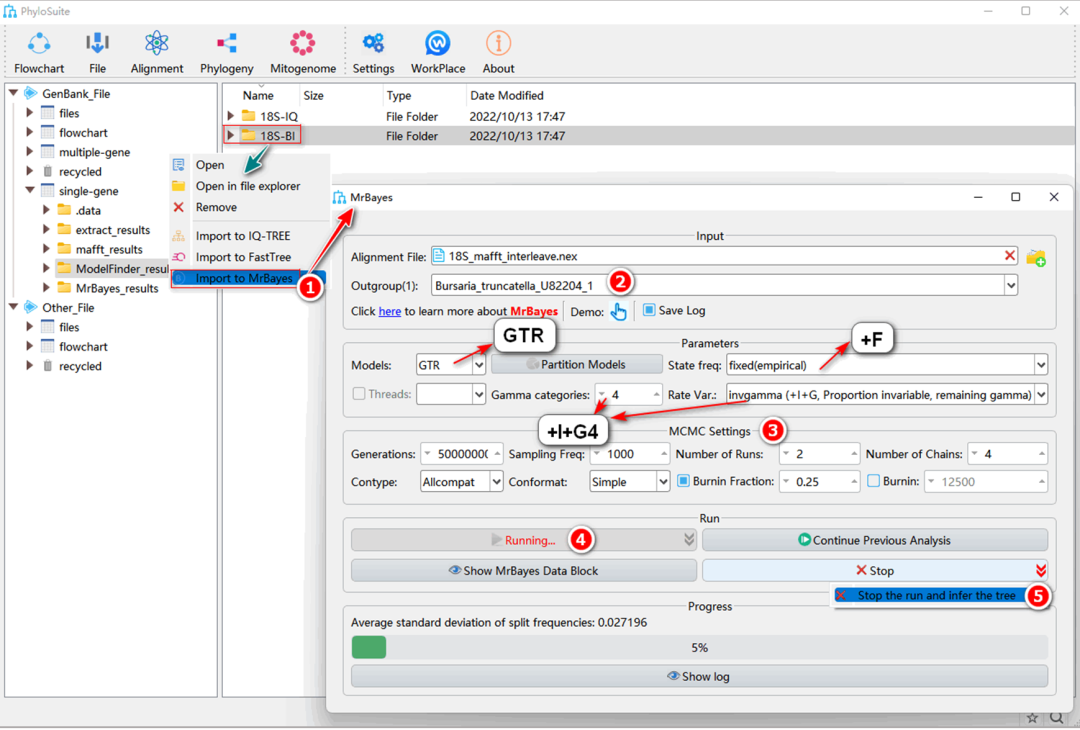
5.6. 重建BI法系统发育树
5.6.1. 右键单击“ModelFinder”的结果文件夹,选择“Import to MrBayes”。MSA文件和最优进化模型(GTR+F+I+G4)将被自动导入。
5.6.2. 选择外群。
5.6.3. “MCMC Settings”请参阅1.11.3。
5.6.4. 参数配置完成后,设置结果文件夹名称(在这里命名为“BI-18S”),然后单击 “Start”按钮开始重建BI法系统发育树。
5.6.5. 当“Average standard deviation of split frequencies”的值低于0.01以及其它指标(例如PSRF和ESS,详见:“如何评估BI运行是否收敛?”)达到收敛状态时,单击“Stop”按钮右侧的下拉菜单并选择“Stop the run and infer the tree”以停止程序并得到结果文件(图S6)。如果结果没有收敛,可以继续运行(详见:“如果BI运行结束但不收敛怎么办?”)。
提示:构建好的BI树保存在“*.con.tre”文件中。
6.使用iTOL美化系统进化树
与本文所述的内置功能(或插件程序)不同,iTOL尚未整合到PhyloSuite中,但PhyloSuite可以生成用于在iTOL中进行系统发育树美化的文件。在对系统发育树美化之前,需先在iTOL网站(https://itol.embl.de)注册登录。
注意:此版本的PhyloSuite附带的内置系统发育树美化功能(Tree annotation)尚未完成,因此本文仅展示如何使用iTOL对系统发育树进行美化。
什么是系统发育树美化?
系统发育树美化是指将各种信息(例如分类学信息,基因组结构,遗传性状等)映射到系统发育树进行可视化的过程。
为什么要美化系统发育树?
系统发育树美化可有助于研究者快速掌握重要的系统发育信息,进而快速准确地识别进化事件。
如何在iTOL中美化系统发育树?
为方便演示,此处使用55种扁形动物的系统发育树进行展示。
6.1. 首先打开iTOL的界面(网址:https://itol.embl.de),登录并选择“My Trees”。
6.2. 在MrBayes的结果文件夹中找到树文件(*.con.tre),然后将文件拖放到“Tree upload”界面中(图S7)。
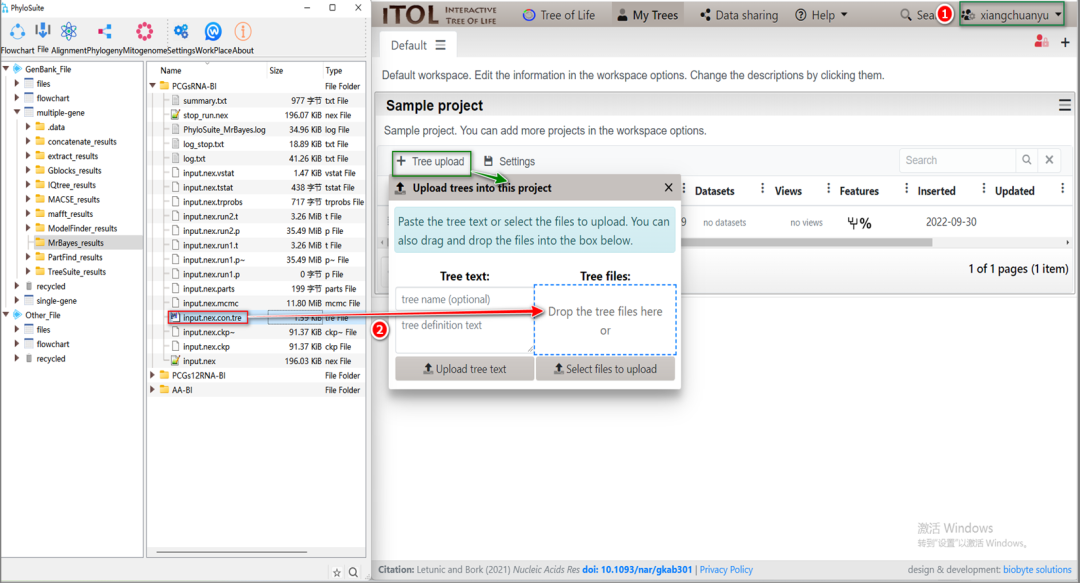
图S7. 将树文件导入iTOL
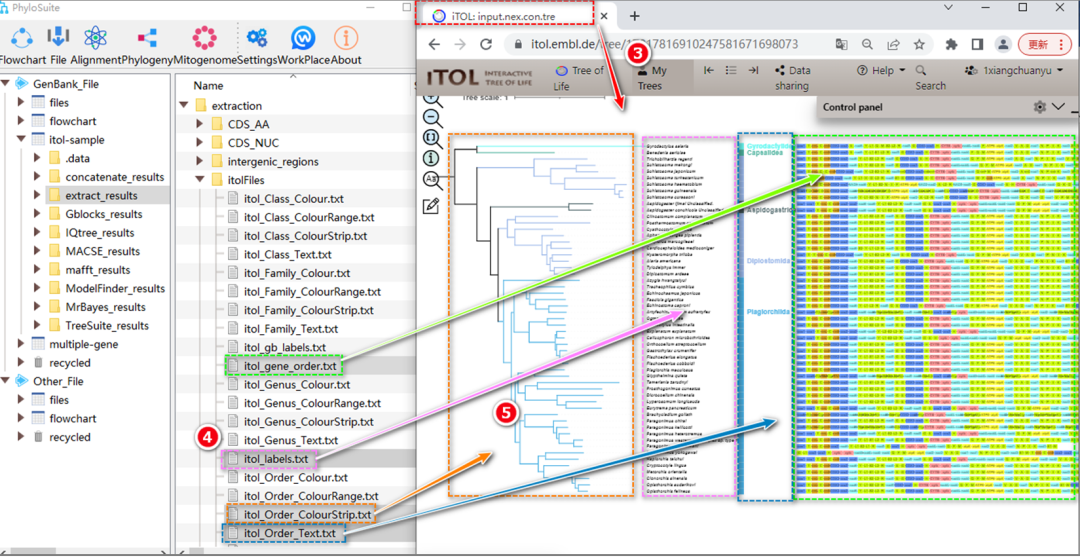
图S8. 使用PhyloSuite生成的iTOL数据集对系统发育树进行美化
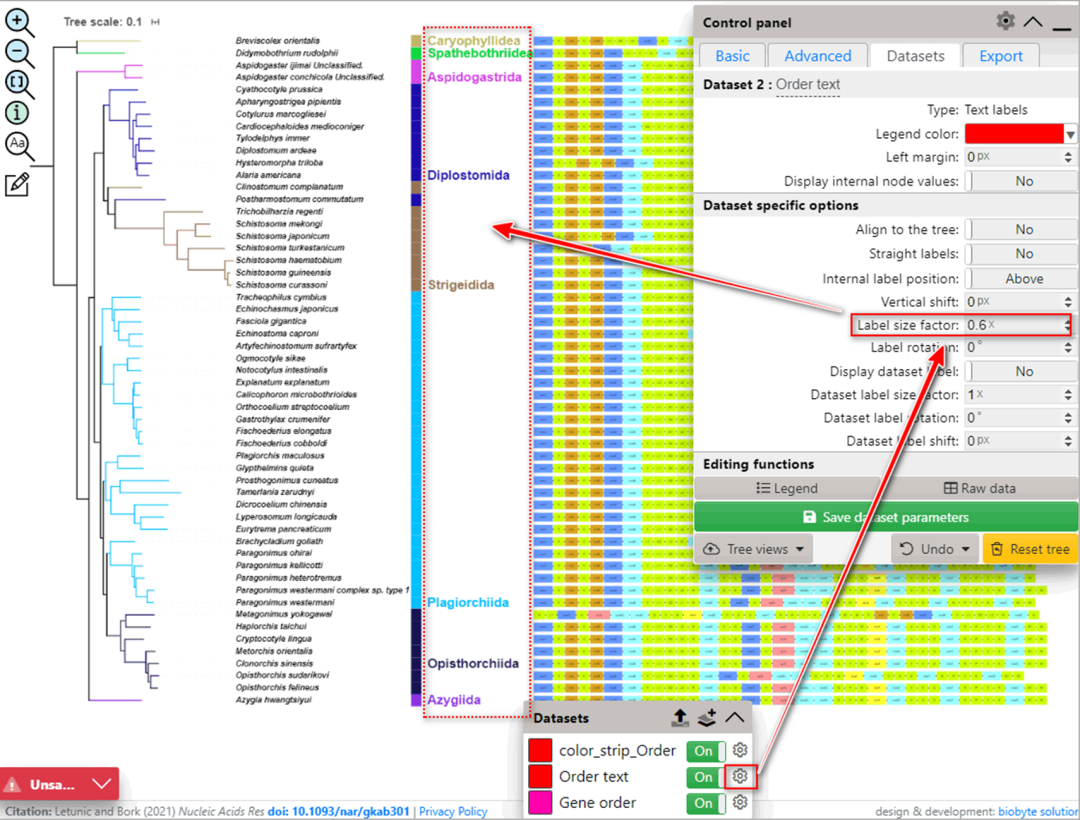
图S9. 调整iTOL的参数
图S10. 在iTOL界面中调整Bootstrap信息
图S11. 导出美化完成的系统发育树
6.3. 双击树文件名称进入树编辑界面。找到“序列提取”的结果文件夹(详见“1.4. 序列提取”部分),“itolFiles”文件夹中的所有文件都可以用于iTOL美化,只需将文件拖到iTOL界面中即可。例如,“itol_labels.txt”文件可用于将物种名称替换为对应的拉丁学名。
6.4. 根据文件名查看它们携带的分类信息。例如“itol_Order_ColourStrip.txt”和“itol_Order_Text.txt”文件可用于注释目的分类信息。
6.4.1. 将两个文件(itol_Order_ColourStrip.txt和itol_Order_Text.txt)一起拖入iTOL界面(图S8)。
6.4.6. iTOL将每个文件视为不同的数据集。可以通过单击“settings”按钮更改每个数据集的参数。
6.4.3. 设置文本数据集的“Text size factor”以更改类名称的字体大小(图S9)。
6.4.4. 设置color_strip数据集的“Strip width”以更改垂直块的宽度。
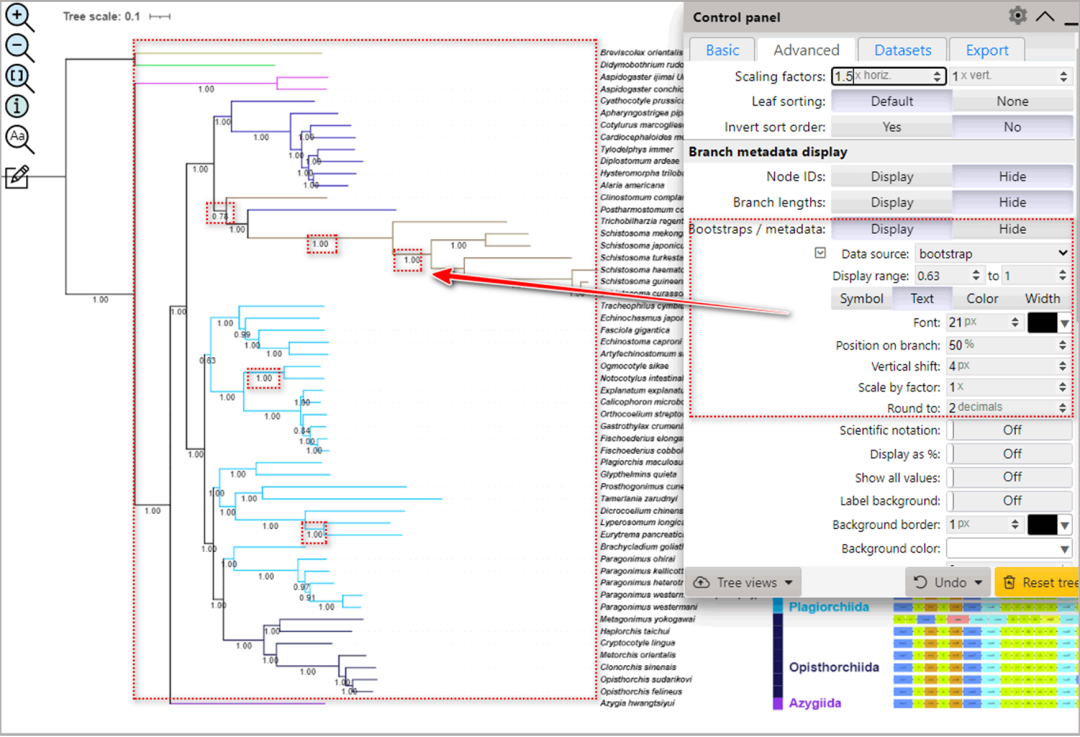
6.4.5. 可以在“Advanced”中的“Bootstraps/metadata”选项中设置Bootstrap值及其相关参数(图S10)。
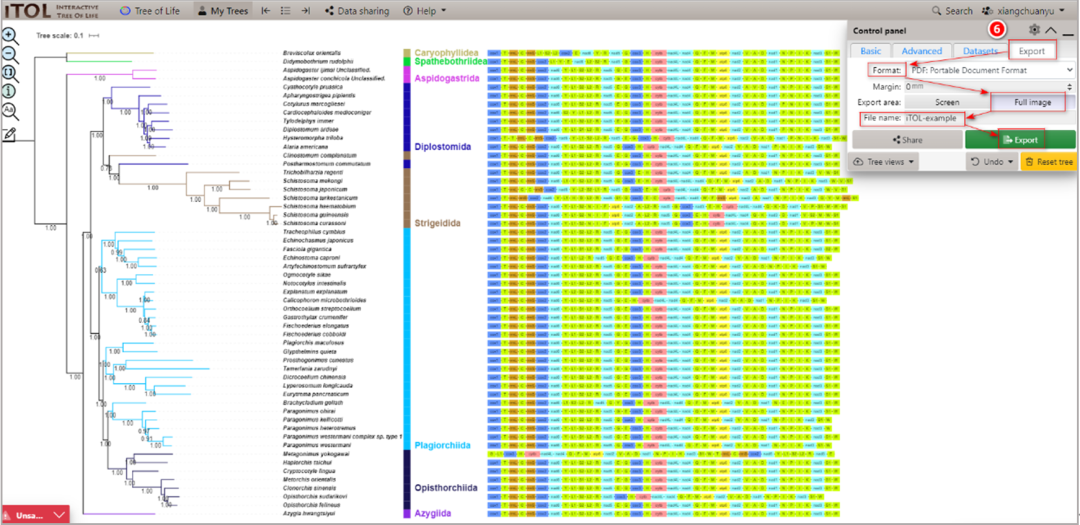
6.5. 系统发育树的美化完成后,单击“Export导出树文件(图S11)。
提示:美化ML法系统发育树(*.treefile)的操作与BI树相同。注意,用于iTOL美化的文件均在“序列提取”的结果文件夹“itolFiles”文件夹中。除此之外,由于在iTOL中保存美化完成的树需要支付费用,因此建议在美化完成后就及时导出(否则退出iTOL界面后,树文件将恢复到原始状态)。
7.输入/输出文件介绍
PhyloSuite及其插件程序仅支持标准文件格式:GenBank、Fasta、Phylip、Nexus等(表S1),输入文件的例子文件详见:http://phylosuite.jushengwu.com/dongzhang0725.github.io/example/。
表S1. PhyloSuite中主要插件的输入/输出文件格式
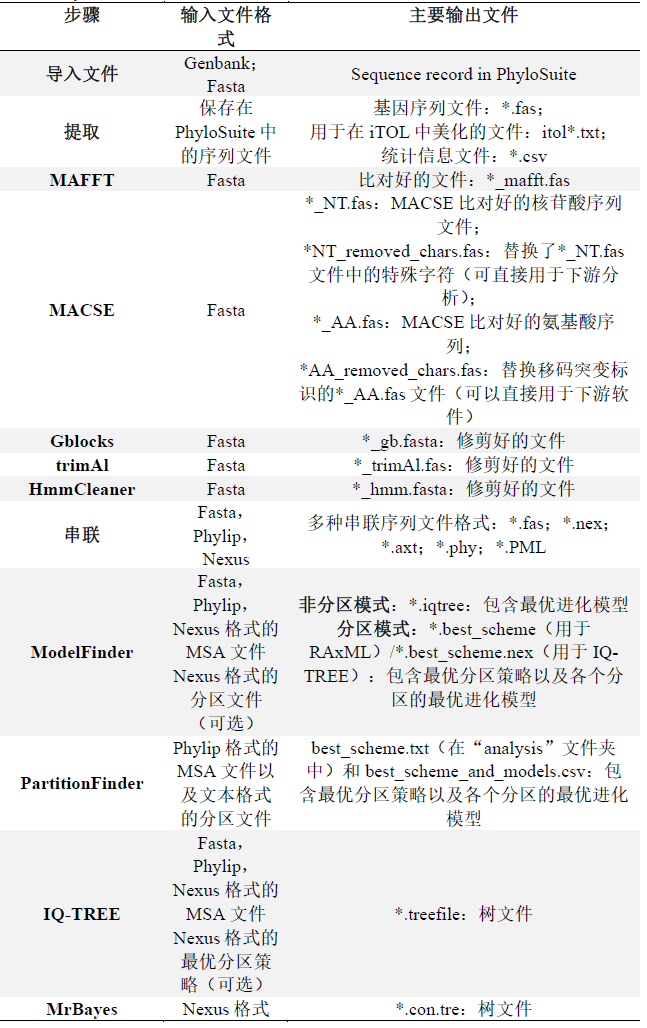
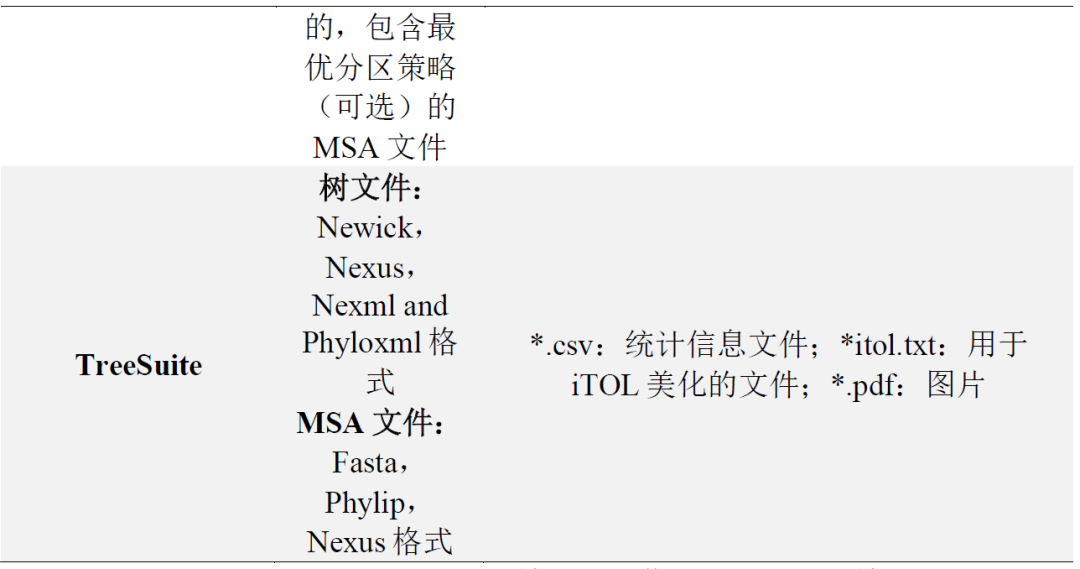
PhyloSuite将自动识别上游程序的输出结果作为下游程序的输入(如自动将MAFFT的运行结果输入到Gblocks中),从而实现流程化运行。以下是某些步骤的输出文件的详细信息。
7.1. 序列提取的输出文件

图S12. 提取步骤的输出文件。选择了“itol-sample”工作文件夹的“extract_results” 结果文件夹进行说明
提取的tRNA、rRNA、PCGs和AA序列均以Fasta格式(*.fas)分别保存在文件夹“tRNA”、“rRNA”、“CDS_NUC”和“CDS_AA”中;用于iTOL美化的文件存储在“itolFiles”文件夹中;有关去除基因拷贝的详细信息存储在“resolve_duplicates”文件夹的“duplicates_resolving_details.csv”文件中(图S12)。
7.2. ModelFinder的输入/输出文件
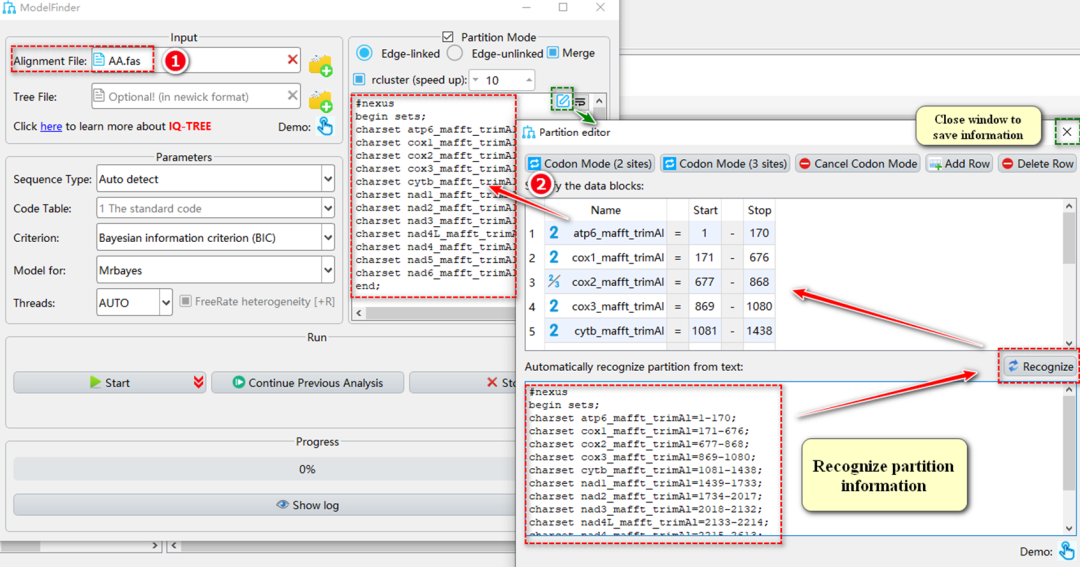
图S13. ModelFinder(分区模式)的输入文件
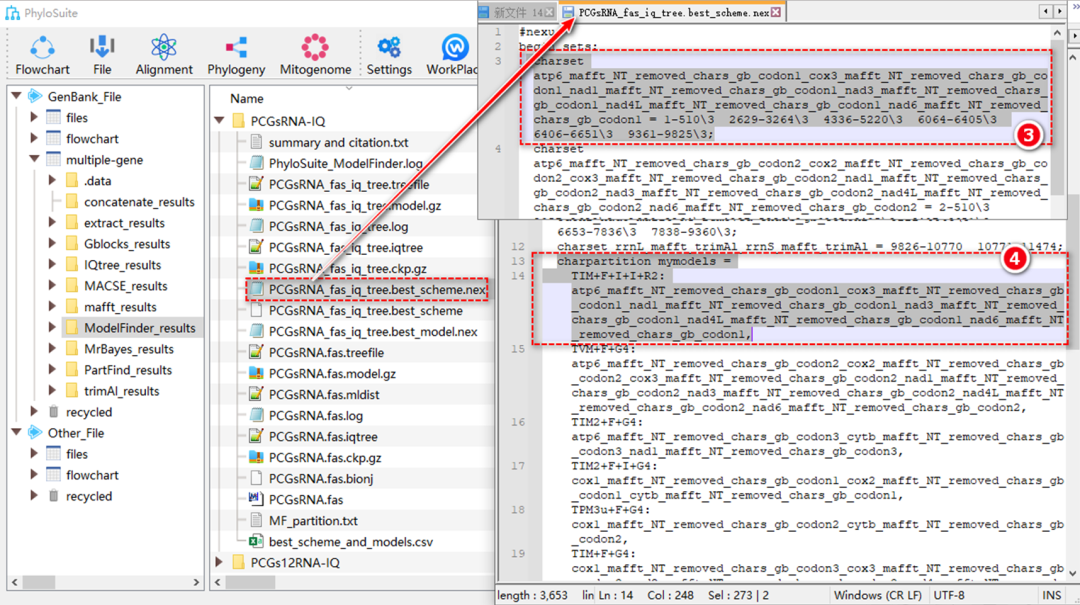
图S14. ModelFinder(分区模式)的输出文件
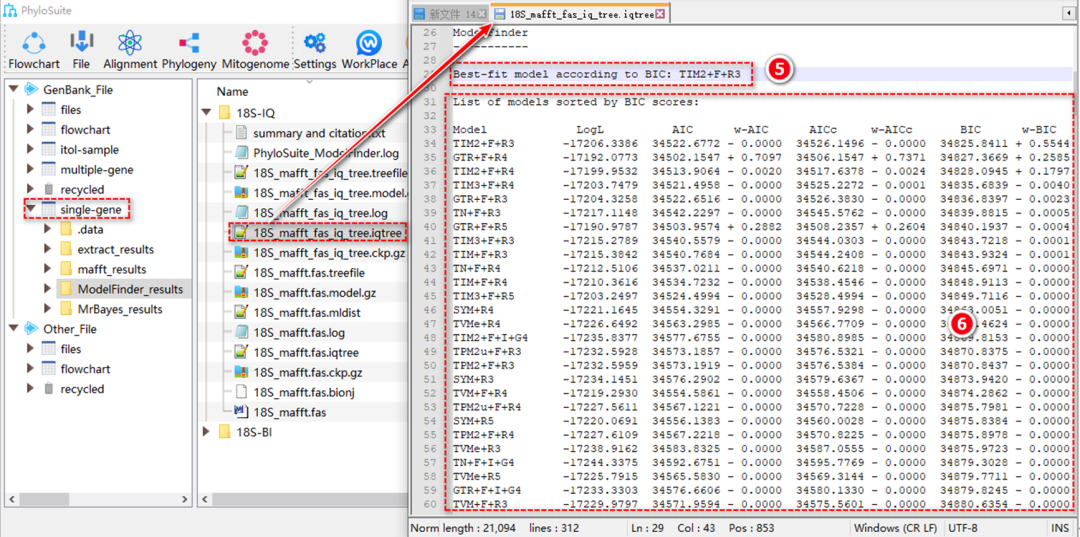
图S15. ModelFinder(非分区模式)的输出文件
7.2.1. 输入MSA文件(*.fas)。
7.2.2. 识别文本格式的预分区信息(图S13)。
7.2.3. 最优分区策略,即划分预分区到不同亚集,并指明它们对应的从开始到结束的位点信息(图S14)。
7.2.4. 每个亚集的最优进化模型(图S14)。
7.2.5. 基于BIC准则选择的最优进化模型(图S15)。
7.2.6. 按BIC分数将模型排序,分数较低模型拟合越好(图S15)。
7.3. PartitionFinder的输入/输出文件
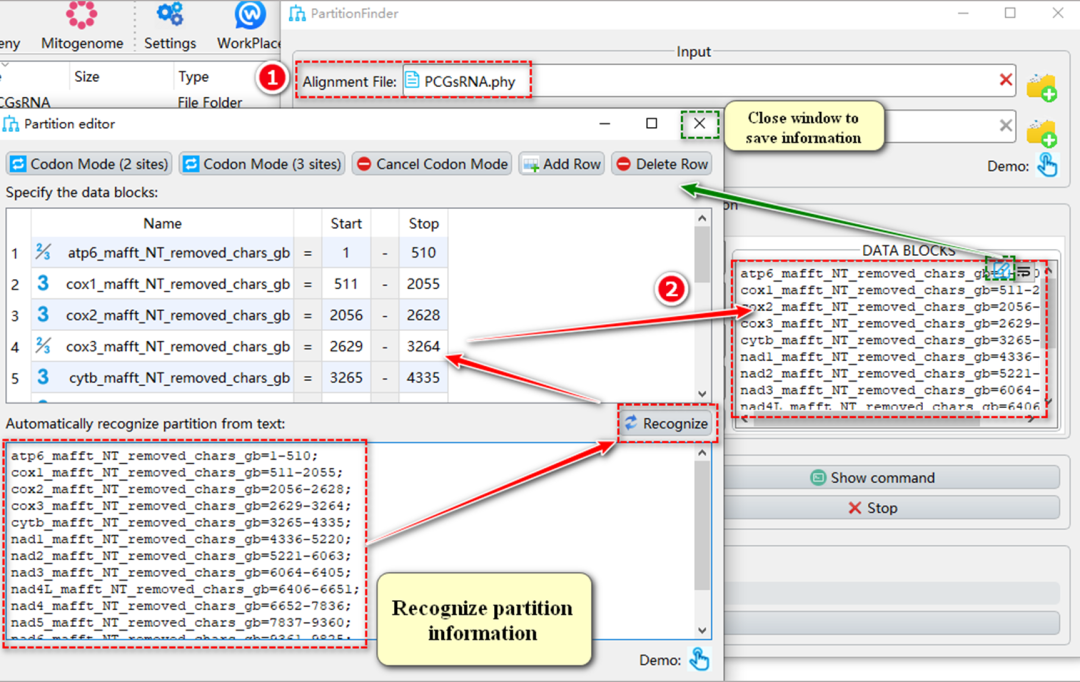
图S16. PartitionFinder的输入文件
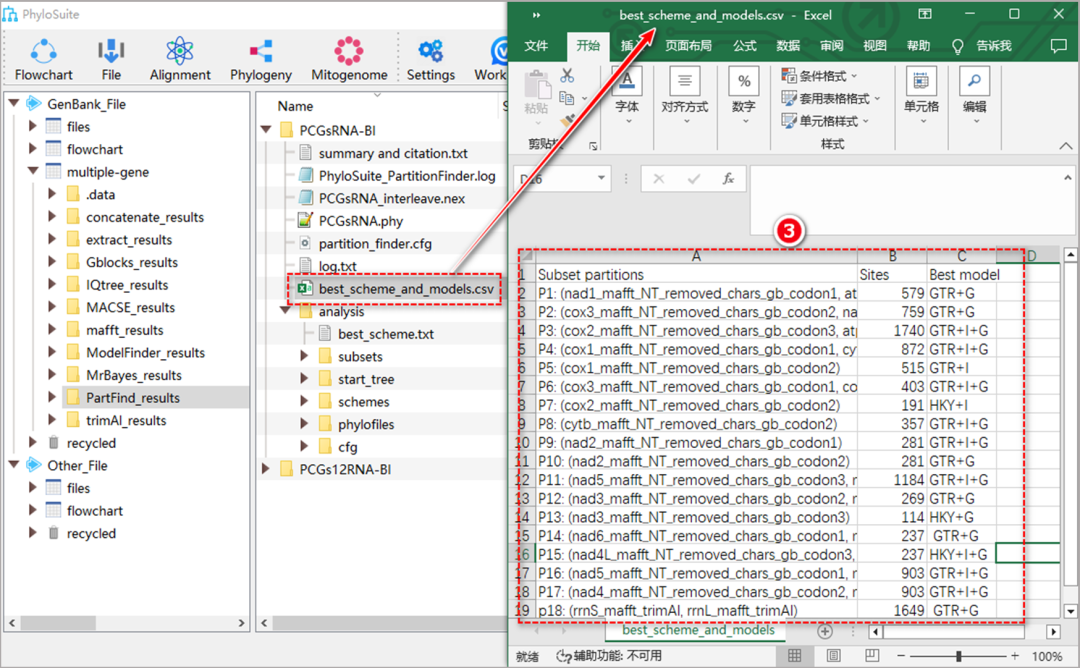
图S17. PartitionFinder的输出文件
7.3.1. 输入MSA文件(*.fas)。
7.3.2. 识别文本格式的预分区信息(图S16)。
7.3.3. “Subset partitions”指最优分区策略;“Sites”指每个亚集中的总位点数;“Best model”指定每个亚集的最优进化模型(图S17)。
7.4. MrBayes的输入文件
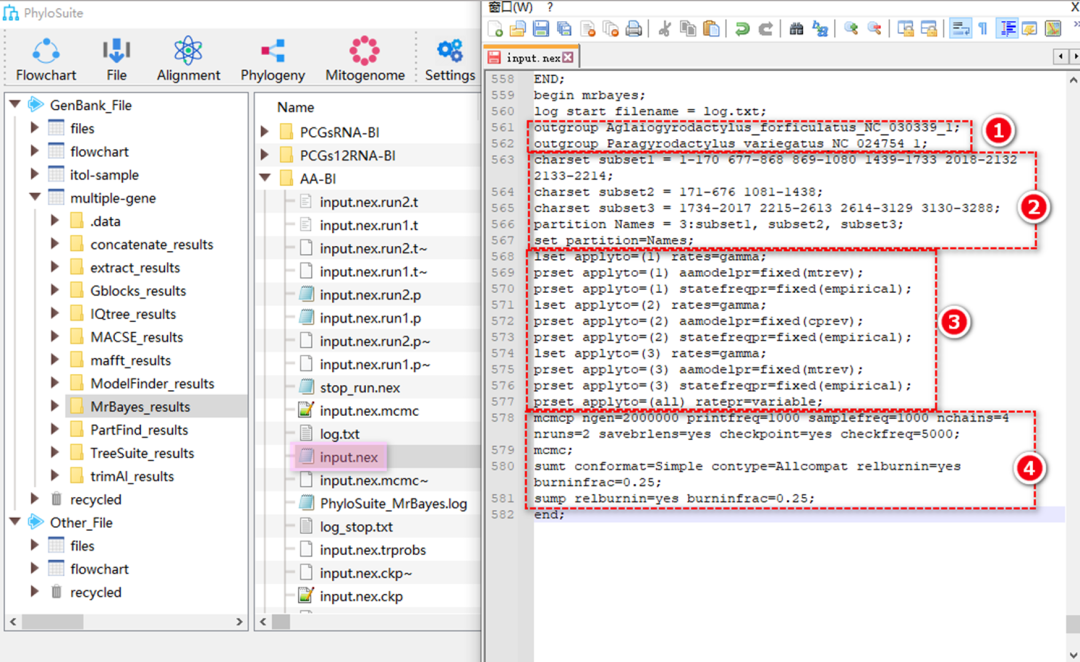
图S18. MrBayes的输入文件
7.4.1. 指定外群。
7.4.2. 指定分区策略信息,包括亚集及其对应的位点。
7.4.3. 指定每个亚集的最优进化模型。
7.4.4. 设置MCMC的参数(图S18),详见第1.11节。
8.故障排除
8.1 输入文件:PhyloSuite只接受标准的文件格式。当使用PhyloSuite的流程化运行功能时,如果用户在结果文件夹中放入了与下游程序所需文件相同后缀的文件,可能会导致PhyloSuite识别错误。
8.2 内存耗尽或工作区文件夹损坏:由于PhyloSuite会永久储存所有运行结果,因此随着时间推移,工作区文件夹会占据较大内存,从而减慢PhyloSuite的运行速度,甚至导致程序崩溃。因此当遇到运行缓慢或崩溃问题,建议首先尝试创建一个新的工作区文件夹,然后重新打开PhyloSuite检查是否工作正常。
8.3 trnL and trnS:在后生动物线粒体基因组的研究中,trnL*和trnS*的模糊注释问题较为常见。线粒体基因组通常编码两个trnL和trnS基因,分别为trnL1和trnL2以及trnS1和trnS2,因此当“序列提取”的结果文件夹的“tRNA”文件夹中有名为“trnL”或“trnS”的FASTA格式文件时,需要对内部的序列进行重新注释,以确定它们具体属于哪一个tRNA。
致 谢
这项工作得到了国家自然科学基金(32102840,31872604)、兰州大学人才引进启动经费(561120206)、甘肃省科技厅项目(21JR7RA533)的支持。感谢周秩健博士提供的技术建议。最后特别感谢广大PhyloSuite用户的反馈和建议,帮助我们不断完善PhyloSuite平台。
数据可用性声明
PhyloSuite(v 1.2.3)对所有研究人员和用户公开开放,安装网址为:http://phylosuite.jushengwu.com/dongzhang0725.github.io/installation/ 或https://dongzhang0725.github.io/installation/
引文格式:
Xiang, Chuan‐Yu, Fangluan Gao, Ivan Jakovlić, Hong‐Peng Lei, Ye Hu, Hong Zhang, Hong Zou, Gui‐Tang Wang, and Dong Zhang. 2023. “Using PhyloSuite for molecular phylogeny and tree‐based analyses.” iMeta. e87. https://doi.org/10.1002/imt2.87
作者简介

相传钰(第一作者)
● 兰州大学硕士研究生在读
● 2022年毕业于曲阜师范大学,同年进入兰州大学生态学院攻读硕士。目前主要研究课题为鱼类寄生虫与宿主的协同进化

张东(通讯作者)
● 兰州大学生态学院/西藏大学理学院青年研究员
● 于2020年博士毕业于中国科学院水生生物研究所,现主要从事鱼类寄生虫的分子系统发育、寄生虫与宿主的协同进化等方面的研究工作,并开发了相关的分析工具,在Molecular Ecology、Molecular Phylogenetics and Evolution、Molecular Ecology Resources、International Journal for Parasitology等生态学、进化生物学、寄生虫学领域的国际知名期刊上发表SCI论文40余篇,1篇入选ESI高被引论文,累计被引1600余次
在线资源
PhyloSuite公众号(发布更新动态、使用技巧、踩坑经验等)
使用交流QQ群

更多推荐
(▼ 点击跳转)
高引文章 ▸▸▸▸
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法
▸▸▸▸
iMeta | 浙大倪艳组MetOrigin实现代谢物溯源和肠道微生物组与代谢组整合分析
▸▸▸▸
iMeta | 高颜值绘图网站imageGP+视频教程合集

第1卷第1期

第1卷第2期

第1卷第3期

第1卷第4期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science