RagTag简介
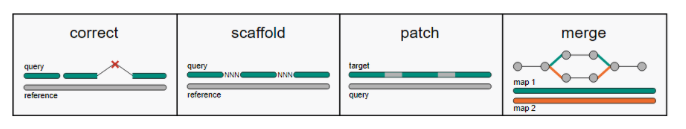
RagTag可以进行错误组装校正、scaffold组装和修补、scaffold合并等,一共分四步:correct,scaffold,patch,merge。之后,可以用Liftoff进行基因注释。
RagTag的conda安装
conda install -c bioconda ragtag1. correct
校正是使用参考基因组来鉴定和校正contigs中的组装错误,该步骤不会将序列减少或增加,仅仅是将序列在错误组装的位置进行打断。
ragtag.py correct relatives-reference.fa scaffolds_FINAL.fasta -t 80结果输出在文件夹./ragtag_output/中。
所有参数:
usage: ragtag.py correct <reference.fa> <query.fa>Homology-based misassembly correction: Correct sequences in 'query.fa' by comparing them to sequences in 'reference.fa'>positional arguments:<reference.fa> reference fasta file (uncompressed or bgzipped)<query.fa> query fasta file (uncompressed or bgzipped)optional arguments:-h, --help show this help message and exitcorrection options:-f INT minimum unique alignment length [1000]--remove-small remove unique alignments shorter than -f-q INT minimum mapq (NA for Nucmer alignments) [10]-d INT maximum alignment merge distance [100000]-b INT minimum break distance from contig ends [5000]-e <exclude.txt> list of reference headers to ignore [null]-j <skip.txt> list of query headers to leave uncorrected [null]--inter only break misassemblies between reference sequences--intra only break misassemblies within reference sequences--gff <features.gff> don't break sequences within gff intervals [null]input/output options:-o PATH output directory [./ragtag_output]-w overwrite intermediate files-u add suffix to unaltered sequence headersmapping options:-t INT number of minimap2/unimap threads [1]--aligner PATH whole genome aligner executable ('nucmer', 'unimap' or 'minimap2') [minimap2]--mm2-params STR space delimited minimap2 whole genome alignment parameters (overrides '-t') ['-x asm5']--unimap-params STR space delimited unimap parameters (overrides '-t') ['-x asm5']--nucmer-params STR space delimted nucmer whole genome alignment parameters ['--maxmatch -l 100 -c 500']validation options:--read-aligner PATH read aligner executable (only 'minimap2' is allowed) [minimap2]-R <reads.fasta> validation reads (uncompressed or gzipped) [null]-F <reads.fofn> same as '-R', but a list of files [null]-T STR read type. 'sr', 'ont' and 'corr' accepted for Illumina, nanopore and error corrected long-reads,respectively [null]-v INT coverage validation window size [10000]--max-cov INT break sequences at regions at or above this coverage level [AUTO]--min-cov INT break sequences at regions at or below this coverage level [AUTO]** The reference and query FASTA files are required **2. scaffold
该步骤是将相邻的contigs序列用100个N连起来,序列的位置和方向需要根据与参考基因组的比对结果确定。
ragtag.py scaffold relatives-reference.fa ragtag_output/ragtag.correct.fasta -t 80 -C ## -C会将没地方放的contig/scaffold连在一起放到chr0中(中间用100个N连接)
最后得到的重要的结果:
ragtag.scaffold.fasta
ragtag.scaffold.agp
插播:
如果除了chr0还有一些碎的contig没放进去,可以用下面的python脚本处理:
#!/public/home/zhangchaofan/anaconda3/bin/python
# -*- coding: utf-8 -*-
##python 02.add_seq.py -s mapped.filter.list.fasta -g test.apg -o test.fa
"""
@File : add_seq.py
@Time : 2021-12-14 22:07:37
@Version : 1.0
@License : (C)Copyright 2020-2021
@Desc : None
@Usage : None
"""
import argparse
from multiprocessing import Pool
import os
import subprocess
import sysdef err_exit():sys.exit('[1;31;47m!!The program exited abnormally, please check the log file !![0m')def Argparse():group = argparse.ArgumentParser()group.add_argument("-i", '--inf', help="Please input total_seq_file!")group.add_argument('-s', '--seqs', help="Please input the to_add_seq")group.add_argument('-g', '--gff', help="please input the gff_file!")return group.parse_args()def main():inf_lines = open(inf).readlines()total_seq = inf_lines[1].strip()# 将要添加的序列读入内存id_seq = {}for line in open(seqs).readlines():if line[0] == '>':# 不保留 > id = line.strip()[1:]else:id_seq[id] = line.strip()sort_id = sorted(id_seq.items(), key=lambda item: len(item[1]), reverse=True)# end_pos = 134326222# 先加N 后加序列gff_lines = open(gff).readlines()end_temp = gff_lines[-1].strip().split()end_pos = int(end_temp[2])end_order = int(end_temp[3]) + 1for tuple_ in sort_id:total_seq += 'N' * 100temp_gff_line = 'Chr0_RagTag\t%d\t%d\t%d\tU\t100\tcontig\tno\tna\n' % (end_pos+1, end_pos+100, end_order)end_order += 1gff_lines.append(temp_gff_line)total_seq += tuple_[1]end_pos += 100temp_gff_line = 'Chr0_RagTag\t%d\t%d\t%d\tW\t%s\t1\t%d\t+\n' % (end_pos+1, end_pos+len(tuple_[1]), end_order, tuple_[0], len(tuple_[1]))gff_lines.append(temp_gff_line)end_order += 1end_pos += len(tuple_[1])gff_ouf_w = open(gff+'.change', 'w')gff_ouf_w.write(''.join(gff_lines))gff_ouf_w.close()seq_ouf = open(inf+'.change', 'w')seq_ouf.write(inf_lines[0]+total_seq+'\n')seq_ouf.close()if __name__ == '__main__':opts = Argparse()inf = opts.infseqs = opts.seqs gff = opts.gffmain()chr0.list.fasta ##已合并chr0的fasta序列文件
contig.list.fasta ##剩下还有的contig的fasta序列文件
运行后contig.list.fasta中的序列会从大到小接在chr0后面,中间用100个N连接,最后生成新的fasta文件和agp文件。
所有参数:
usage: ragtag.py scaffold <reference.fa> <query.fa>Homology-based assembly scaffolding: Order and orient sequences in 'query.fa' by comparing them to sequences in 'reference.fa'positional arguments:<reference.fa> reference fasta file (uncompressed or bgzipped)<query.fa> query fasta file (uncompressed or bgzipped)optional arguments:-h, --help show this help message and exitscaffolding options:-e <exclude.txt> list of reference sequences to ignore [null]-j <skip.txt> list of query sequences to leave unplaced [null]-J <hard-skip.txt> list of query headers to leave unplaced and exclude from 'chr0' ('-C') [null]-f INT minimum unique alignment length [1000]--remove-small remove unique alignments shorter than '-f'-q INT minimum mapq (NA for Nucmer alignments) [10]-d INT maximum alignment merge distance [100000]-i FLOAT minimum grouping confidence score [0.2]-a FLOAT minimum location confidence score [0.0]-s FLOAT minimum orientation confidence score [0.0]-C concatenate unplaced contigs and make 'chr0'-r infer gap sizes. if not, all gaps are 100 bp-g INT minimum inferred gap size [100]-m INT maximum inferred gap size [100000]input/output options:-o PATH output directory [./ragtag_output]-w overwrite intermediate files-u add suffix to unplaced sequence headersmapping options:-t INT number of minimap2/unimap threads [1]--aligner PATH aligner executable ('nucmer', 'unimap' or 'minimap2') [minimap2]--mm2-params STR space delimited minimap2 parameters (overrides '-t') ['-x asm5']--unimap-params STR space delimited unimap parameters (overrides '-t') ['-x asm5']--nucmer-params STR space delimted nucmer parameters ['--maxmatch -l 100 -c 500']** The reference and query FASTA files are required **3. patch
该步骤是用contigs序列对上一步得到的scaffold序列进行gap填补。该步骤比较耗时,如果急需使用基因组进行后续分析,可以省略该步骤。
ragtag.py patch ./ragtag_output/ragtag.scaffold.fasta scaffolds_FINAL.fasta -t 80以上均已亲测。
4. merge
在scaffolding过程中,可能会根据不同参数或图谱数据产生多个版本的基因组组装结果,该步骤可以将多个结果根据权重进行最终组装结果的生成。
如果有HiC数据,还可以加入HiC数据生成比较好的组装结果。
参考来源:
基于参考基因组的基因组组装和注释 - 简书
GitHub - malonge/RagTag: Tools for fast and flexible genome assembly scaffolding and improvement