目录
- 1. 设置阈值来显示对应的基因名
- 2. 指定基因名展示
写在前面——之前写的RNA-seq(一到四)是根据别人文章中提到的数据进行一系列分析的,但是找公司做的单细胞测序,一般不需要自己进行数据清洗之类的操作,公司会直接给个clean_data,以及所有的你需要的文件,或者一个云系统的账号。所以我们最终要做的就是根据这些数据,来绘制达到文章发表级别的图,来说明我们实验想表达的事情。
注:本文虽然是RNA-seq——五,但与前几个使用的并不是同一个数据集,本文数据集是私有数据集。
参考:
给火山图上标记基因名字
火山图|给你geneList,帮我标到火山图上
多种方法在火山图上标记感兴趣基因(差异基因,或者通路)
1. 设置阈值来显示对应的基因名
library(readxl)
library(ggrepel)# 读取差异基因数据集
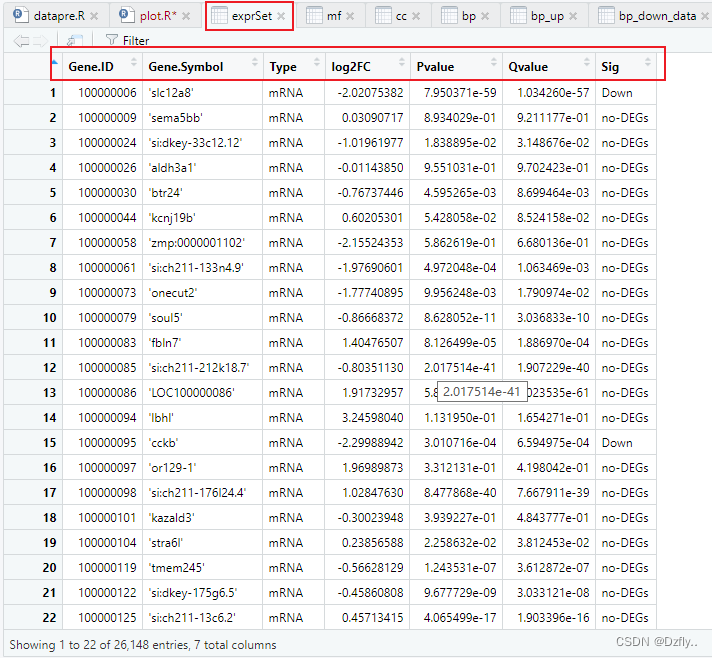
exprSet <- read_xlsx("allgene.xlsx")
colnames(exprSet) <- c("Gene ID", "Gene Symbol", "Type", "log2FC", "Pvalue", "Qvalue")# 设置阈值,整理数据
# 阈值不同,结果不同
cut_off_qvalue = 0.01
cut_off_logFC = 2
exprSet$Sig <- ifelse(exprSet$Qvalue < cut_off_qvalue & abs(exprSet$log2FC) >= cut_off_logFC, ifelse(exprSet$log2FC > cut_off_logFC ,'Up','Down'),'no-DEGs')exprSet <- data.frame(exprSet)
# tmp <- tmp %>% drop_na(Sig)
table(exprSet$Sig)ggplot(exprSet, aes(x = log2FC, y = -log10(Qvalue), colour=Sig)) +geom_point(alpha=0.4, size=3.5) +scale_color_manual(values=c("#546de5", "#d2dae2","#ff4757")) + xlim(c(-16, 16)) + # 辅助线geom_vline(xintercept=c(-cut_off_logFC,cut_off_logFC),lty=4,col="black",lwd=0.8) +geom_hline(yintercept = -log10(cut_off_qvalue),lty=4,col="black",lwd=0.8) +# 坐标轴labs(x="Fold Change", y="-log10 (Q-value)") +# 主题theme_bw() +# 标题ggtitle("Q-value vs Fold Change") +# 图例theme(plot.title = element_text(hjust = 0.5), legend.position="right", legend.title = element_blank() ) + # 给点标上基因名geom_text_repel(# 可以设置跟上面不同的阈值,用数值替换即可data = subset(exprSet, exprSet$Qvalue < cut_off_qvalue & abs(exprSet$log2FC) >= cut_off_logFC),aes(label = Gene.Symbol), size = 3,box.padding = unit(0.5, "lines"),point.padding = unit(0.8, "lines"), segment.color = "black", show.legend = FALSE )
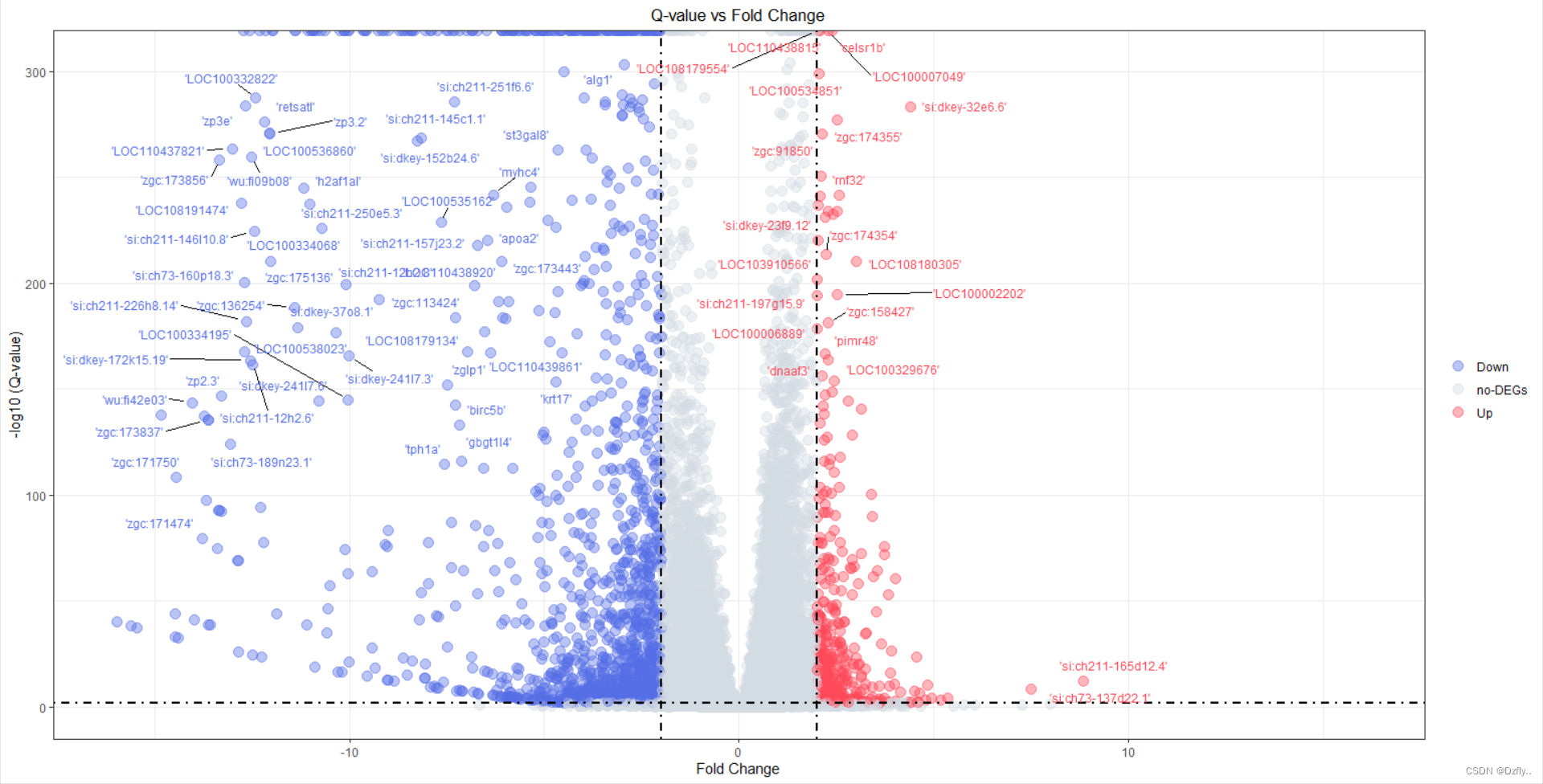

可以看到,有一千八百多个基因名无法显示,这里我们可以把qvalue和flod change设置的再严格一些,或者直接指定一些基因来标注。
2. 指定基因名展示
library(dplyr)
library(ggrepel)# 将geneList中的基因全部标注
# my_label <- label_1
# gene <- my_label$`Gene Symbol`
# gene <- data.frame(gene)
# gene$geneList <- gene$gene
# tmp <- exprSet %>% left_join(gene,by = c("Gene.Symbol" = "gene"))# 简单粗暴的标注
gene_tmp <- c("'cfd'", "'c9'", "'fga'", "'c3a.1'", "'fgg'","'il2rga'", "'ccl25b'", "'cxcl8b.3'", "'xcr1b.2'", "'ccl20a.3'","'mhc2dab'", "'col1a2'", "'gna14'", "'akt1'", "'fcer1g'","'ptgdsb'", "'ptgdsb.2'", "'ggt1a'", "'pla2g12b'", "'ptges'", "'ptgs2a'","'il1b'")
gene_tmp <- data.frame(gene_tmp)
gene_tmp$geneList <- gene_tmp$gene_tmptmp <- exprSet %>% left_join(gene_tmp,by = c("Gene.Symbol" = "gene_tmp"))ggplot(tmp, aes(x = log2FC, y = -log10(Qvalue), colour=Sig)) +geom_point(alpha=0.4, size=3.5) +scale_color_manual(values=c("#546de5", "#d2dae2","#ff4757")) + xlim(c(-16, 16)) + # 辅助线geom_vline(xintercept=c(-cut_off_logFC,cut_off_logFC),lty=4,col="black",lwd=0.8) +geom_hline(yintercept = -log10(cut_off_qvalue),lty=4,col="black",lwd=0.8) +# 坐标轴labs(x="Fold Change",y="-log10 (Q-value)")+theme_bw()+ggtitle("Q-value vs Fold Change")+# 图例theme(plot.title = element_text(hjust = 0.5), legend.position="right", legend.title = element_blank() ) + geom_label_repel(aes(label=geneList), fontface="bold",color="grey50", box.padding=unit(0.35, "lines"),point.padding=unit(0.5, "lines"), segment.colour = "grey50")
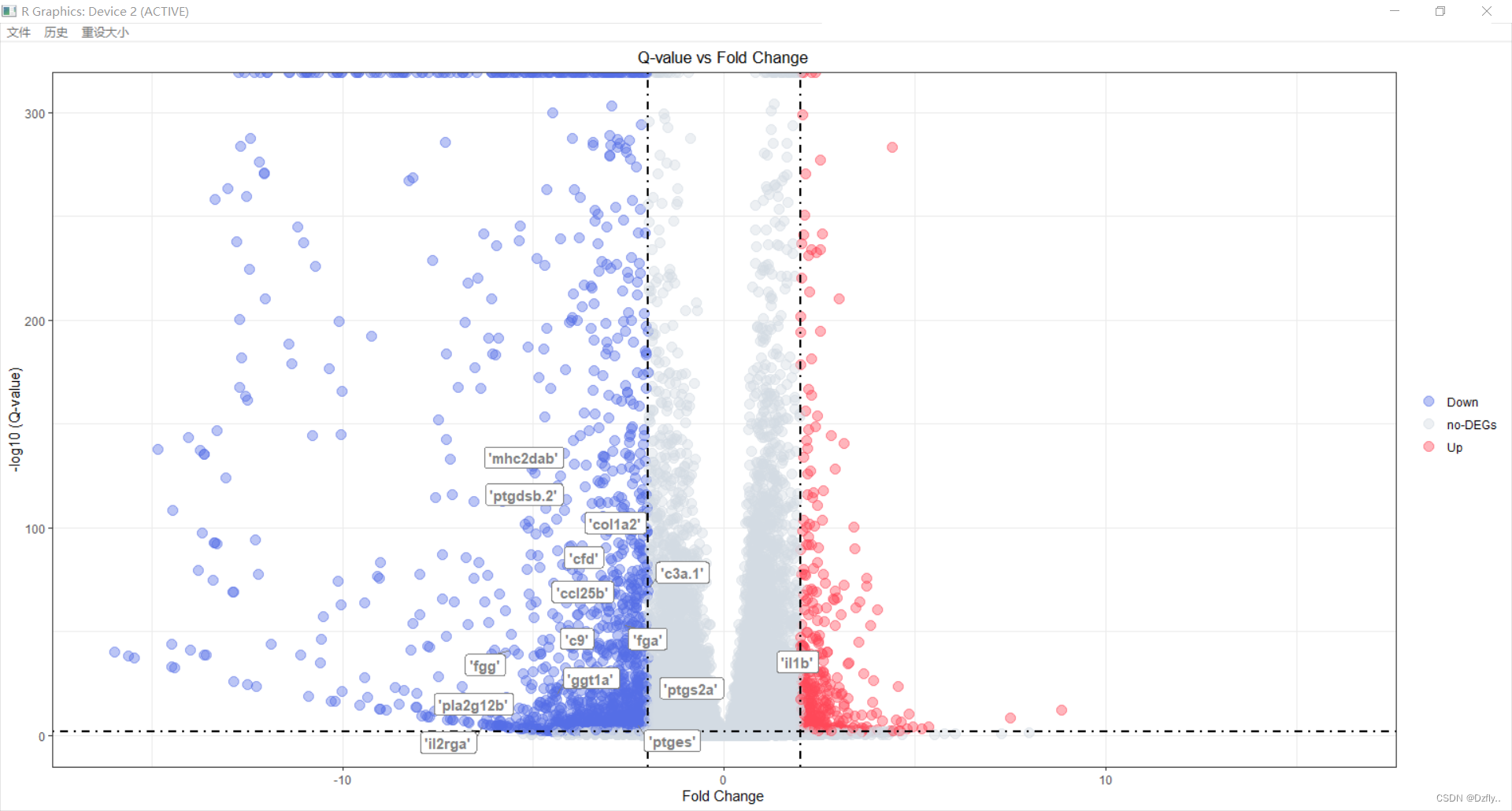

竟然有几个不显示,估计字体设置的太大了,挤不下。方法掌握,之后就可以自己慢慢的去调数值了。
![教程 | 规模化物种同源基因分析 - orthofinder [上篇]](https://img-blog.csdnimg.cn/img_convert/0d3225553fbefb5bcbef1fb39e9cf55e.png)